

1. 서 론
2. 분자동역학 해석 방법
3. 결과 및 토의
3.1 분해 시뮬레이션 결과
3.2 주요 생성물 선택도에 대한 개시제의 영향
3.3 PAH 전구체 생성에 대한 개시제의 영향
3.4 개시제 종류에 따른 연료의 반응 경로 메커니즘 분석
4. 결 론
기호설명
CHP : Cumene hydroperoxide
DADP : Diacetone diperodixe
DEE : Diethyl ether
DFT : Density functional theory
DTBP : Di-tert-butyl peroxide
exo-THDCPD : exo-tetrahydrodicyclopentadiene
HCF : Hydrocarbon fuel
MCH : Methylcyclohexane
MTBE : Methyl tert-butyl ether
NM : Nitromethane
PAH : Polycyclic aromatic hydrocarbon
PH : Phenyl hydrazine
PPAMAM : Palmitoyl-poly-(amidoamine)
ReaxFF : Reactive force field
TBA : Tributylamine
TEA : Triethylamine
TEMPO : 2,2,6,6-tetramethyl-1-piperidinyloxy
Trigonox : 301 3,6,9-triethyl-3,6,9-trimethyl-1,2,4,5,7,8-hexaoxonane
1-NP : 1-nitropropane
1. 서 론
극초음속 비행체는 마하 5 이상의 속도로 지구 대기권 내에서 비행하는 고속 비행체로, 군사적인 목적뿐만 아니라 우주 발사체의 추진 시스템에도 활용될 수 있다. 그러나 이러한 비행체의 개발 과정에서는 여러 기술적인 문제들이 수반된다. 특히, 대기권 내에서 고속 비행 시 동체에 가해지는 공력 가열과 스크램제트 엔진의 연소실에서 발생하는 고온 가열로 인해 비행체의 구조 변형이나 엔진 정지가 발생할 수 있다[1]. 따라서 이러한 고온 열부하 문제를 효과적으로 관리하기 위해서는 냉각 기술 개발이 필수적이다.
국내외에서 극초음속 비행체의 내외부에서 발생하는 고온 가열 문제를 해결하기 위한 재생냉각 방식이 활발히 연구되고 있다[2,3,4]. 재생냉각 기술은 연료가 열을 흡수함으로써 비행체의 고온 부위를 냉각하는 방식이며, 이를 위해 극저온 수소나 액체 탄화수소 연료를 활용하여 효율적인 냉각 효과를 얻을 수 있다. 극저온 수소 연료는 높은 흡열 성능을 지니고 있으나, 부피당 에너지 밀도가 매우 낮고, 저장 효율성도 떨어져 극초음속 비행체와 같은 부피가 제한되는 시스템에는 적용이 어려운 한계가 있다. 반면, 액체 탄화수소 연료는 높은 부피당 에너지 밀도를 지녀 효율적인 저장이 가능하여 실용적인 극초음속 비행체에 적합하다[4]. 그러나 액체 탄화수소 연료는 극저온 수소 대비, 냉각 성능에 한계가 있다. 따라서 이러한 한계를 극복하고 액체 탄화수소 연료의 흡열 성능을 향상시키는 연구가 필수적이다[5,6,7,8].
탄화수소 연료는 재생냉각 채널 내의 고온, 고압 조건에서 열분해되어, 고속 비행 시 요구되는 흡열 성능을 충족할 수 있다. 그러나 이 과정에서 생성되는 불포화 탄화수소는 축합 및 중합 반응을 통해 코크(coke)로 전환된다[5]. 코크는 재생냉각 채널 내에서 연료의 유동을 방해하고, 채널 관벽에 증착되어 열전달 특성을 저하시키는 주요 요인으로 작용한다. 일반적으로 코크의 생성은 온도와 연료의 체류시간에 지수적으로 영향을 받는다[6]. 따라서 코크 생성을 저감하기 위해서는 연료의 열분해 온도 범위를 낮추는 것이 중요하다. 이를 위한 방법으로 화학 개시제를 액체 탄화수소 연료에 첨가하는 연구가 활발히 진행되고 있다.
개시제의 작용 원리를 설명하기에 앞서, 순수 탄화수소 연료의 열분해는 C-C(탄소-탄소) 또는 C-H(탄소-수소) 결합의 해리 반응을 통해 시작된다. 이때 요구되는 해리 에너지는 각각 82~87 kcal/mol(C-C 결합) 및 95 kcal/mol (C-H 결합)으로 높은 편이다. 따라서 순수 연료의 분해 반응을 위해서는 고온의 반응 조건이 필요하다. 하지만 화학 개시제는 상대적으로 낮은 결합 해리 에너지의 C-N(탄소-질소, 64~73 kcal/mol) 또는 C-O(탄소-산소, 71 kcal/mol)를 포함하고 있기 때문에, 순수 연료보다 더 낮은 온도 영역에서 분해되어 라디칼을 생성시킨다[9,10,11]. 이러한 라디칼은 수소 추출 반응(H-abstraction)과 같은 2차 반응(secondary reaction)을 통해 연료의 분해 반응을 가속 시킨다[12].
최근 보고된 화학 개시제 첨가 탄화수소 연료 연구의 주요 사례와 온도 조건은 Table 1에 정리되어 있다. Jia 등[11]은 1-NP 주입을 통해 n-decane의 초기 분해 온도를 낮추고 C2H4 및 C3H6의 선택도를 크게 증가시키는 것을 확인하였다. Kalyen 등[13]은 TBA를 첨가한 HCF-1 연료의 초임계 열분해 실험을 통해, 개시제가 흡열량과 연료 전환율을 증가시킴을 보고하였다. Priyadarshi 등[14]은 Jet-A1 연료에 다양한 종류의 화학 개시제를 첨가하여 열분해 실험을 수행하였다. 실험 결과, TEMPO가 저분자량 알켄 생성에 가장 효과적임을 확인하였고, PH와 1-NP도 마찬가지로 높은 알켄 분율을 보였다. He 등[15]은 PPAMAM를 첨가하여 MCH 열분해 실험을 수행한 결과, 흡열량이 증가하고 연료 전환율이 향상됨을 확인하였다.
이처럼 다양한 실험적 연구는 개시제가 열분해 성능을 향상시키는 데 효과적임을 보였다. 하지만, 향상 메커니즘에 대한 분자 단위에서의 연구는 부족한 실정이다. 특히, 초기 라디칼 생성 및 주요 중간체 생성과 같은 미시적 반응 메커니즘을 실험적으로 규명하는 데는 한계가 있다. 이러한 한계를 보완하기 위해 ReaxFF 분자동역학 시뮬레이션이 활용되고 있다. 그 예로써, Chen 등[16]은 ReaxFF 분자동역학 시뮬레이션과 실험을 비교하여 1-NP가 첨가된 n-hexane의 분해 메커니즘을 규명하였다. Wang 등[17]은 ReaxFF 분자동역학 시뮬레이션을 통해 n-decane에 DEE, MTBE, 1-NP, Trigonox 301, TEA, DADP를 첨가하여 분해 과정을 분석하였다. 시뮬레이션 결과, 개시제의 화학 구조에 따라 생성되는 라디칼이 달라진다. 이에 따라 n-decane의 분해 경로와 반응 속도가 변화되는 것을 확인했다. Guan 등[18]은 n-heptane과 MCH의 분해 과정을 ReaxFF 시뮬레이션을 통해 분석하였으며, NM 첨가에 따른 반응 경로 변화, 라디칼 생성 및 주요 중간생성물의 반응 과정을 평가하였다.
본 연구에서는 ReaxFF 분자동역학 시뮬레이션을 이용하여 고에너지 액체 연료 exo-THDCPD의 분해 메커니즘과 반응 동역학에 대한 화학 개시제의 영향을 분석하였다. 가속 해석 조건(2000 K이상)에서 사전 스크리닝 결과를 바탕으로 총 3종(TEA, DTBP, CHP)의 화학 개시제를 선정하였다. 선정된 개시제를 활용하여 연료의 전환율과 반응 속도에 미치는 영향을 정량적으로 평가하고, 주요 생성물의 선택도를 분석하여 분해 특성의 변화를 규명하였다. 또한, 반응 경로 분석을 통해 개시제 분해 과정에서 생성된 라디칼이 연료의 분해 반응에 미치는 영향을 분석하고, 개시제에 따른 반응 메커니즘을 상세하게 비교하였다. 본 연구 결과는 연료 첨가제를 활용한 연료의 분해 반응 최적화 및 열관리 성능 향상을 위한 기초 자료로 활용될 수 있을 것으로 기대된다.
Table 1.
Examples of previous studies on the effects of various chemical initiators on fuel decomposition. Note that Exp. and Sim. stand for experimental and simulation work, respectively
| Fuel | Chemical initiator | Method | Temperature (K) | Ref. |
| n-decane | 1-NP | Exp. | 323-960 | [11] |
| HCF-1 | TBA | Exp. | 823-953 | [13] |
| Jet-A1 | TEA, TEMPO, PH, 1-NP, DTBP, CHP | Exp. | 863-1193 | [14] |
| MCH, kerosene | PPAMAM | Exp. | 873-993 | [15] |
| n-hexane | 1-NP | Exp. and Sim. |
Exp.: 1103 Sim.: 1103-3103 | [16] |
| n-decane |
DEE, MTBE, 1-NP, Trigonox 301, TEA, DADP | Sim. | 2200-3000 | [17] |
| n-heptane, MCH | NM | Sim. | 2400-3200 | [18] |
2. 분자동역학 해석 방법
본 연구에서 사용한 ReaxFF 분자동역학 시뮬레이션은 연료 분자의 원자간 결합 및 해리 과정을 모사할 수 있어, 개시제와 연료 간의 화학 반응 경로를 예측하는 데 있어 매우 유용하다[18]. Fig. 1은 본 연구에서 사용한 exo-THDCPD 연료와 개시제 3종(TEA, DTBP, CHP)의 분자 구조를 보여주며, 순수 연료와 DTBP가 포함된 연료의 초기 배치 구조를 나타낸다. 28.51 Å × 28.51 Å × 28.51 Å 크기의 계산 영역(simulation box) 내에 40개의 exo-THDCPD 연료 분자를 균일하게 배치하고 밀도가 0.39 g/cm3가 되도록 설정하였다. 개시제가 포함된 혼합물의 경우, 40개의 연료 분자에 3개의 개시제 분자를 주입하였고, 동일한 밀도 조건을 유지하기 위해 계산 영역을 조정하였다. 본 연구에서 고려한 해석 조건에서는 밀도 변화가 연료 분해 과정에 미치는 영향이 거의 없었다.
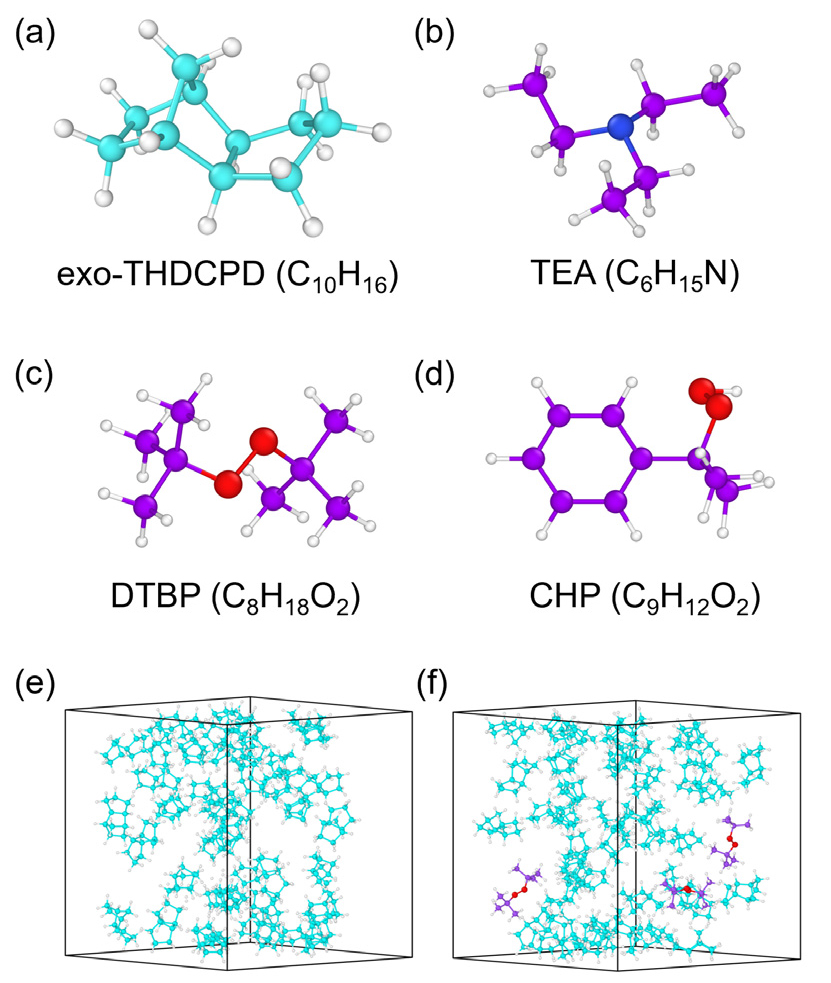
초기 시스템에서 분자 간 에너지를 최소화하기 위해 5 K에서 5 ps 동안 시뮬레이션을 수행한 후, 5개의 초기 시스템을 생성하였다. 이후, 각 시스템을 1500 K에서 10 ps 동안 시뮬레이션하여 평형상태에 도달시킨 후, 1600, 1700, 1800 K의 온도 조건에서 각각 2 ns 동안 시뮬레이션을 수행하였다. 이러한 시뮬레이션 결과를 평균하여 전환율, 반응 속도, 그리고 개시제가 흡열연료의 분해 반응에 미치는 영향을 분석하였다. 모든 시뮬레이션은 NVT 앙상블에서 수행되었으며, 계산 속도를 높이고 비용을 줄이기 위해 시간 간격(time step)은 0.2 fs를 적용하였다[19]. 온도 제어 모델은 Berendsen 방법을 사용하였고, 감쇠 상수는 200 fs를 설정하였다. 본 연구에서 사용한 ReaxFF 힘장은 CHON-2019 모델이다[20]. 이 모델은 기존 CHON-2010[21]를 보완한 힘장 모델로서, C-N 결합의 형성과 해리를 포함한 결합 특성이 DFT 기반 제일원리 계산과 더 높은 일치도를 보인다.
Table 2에는 시뮬레이션 조건을 요약하였다. 앞서 언급했듯이, 통계적 불확실성을 고려하기 위해 모든 계산은 다섯 번의 시뮬레이션을 수행한 후 앙상블 평균을 적용하였다. 통계적 불확도는 95% 신뢰 수준에서 스튜던트 t 분포를 사용하여 추정되었으며, 활성화 에너지의 최대 불확도는 2.97%로 계산되었다. 선택도(selectivity)는 특정 생성물이 반응 과정에서 차지하는 상대적 비율을 나타내는 지표이다. 이러한 선택도를 통해 연료가 주요 생성물로 변환되는 경향을 정량적으로 분석할 수 있다. 본 연구에서 사용한 선택도 계산식은 식 (1)과 같다.
여기에서 는 특정 생성물()의 개수이며, 은 반응하여 소모된 연료 분자의 개수이다.
3. 결과 및 토의
3.1 분해 시뮬레이션 결과
Fig. 2는 순수 연료와 각 화학 개시제의 종류에 따라 1600, 1700, 1800 K의 온도 조건에서 시간에 따른 전환율을 나타낸다. 연료의 전환율은 화학 반응을 통해 연료가 분해된 정도를 나타낸다. 순수 연료는 1600 K에서 초기 반응 동안 열분해가 거의 일어나지 않아 전반적으로 낮은 전환율을 보인다. 반면, CHP와 TEA를 첨가한 경우 낮은 반응 온도에서도 순수 연료 대비 약 2배 높은 전환율을 보였다. 1800 K 조건에서는 전환율이 각각 28%와 11% 상승했다. DTBP를 첨가했을 때, 반응 초기에 순수 연료보다 전환율이 유의미하게 증가하였다. 하지만, 반응 종료 시점에서는 CHP와 TEA보다 낮은 전환율을 보였다. 이는 DTBP가 초기 분해 이후로는 탄화수소 연료의 분해 반응에 참여하지 않기 때문이다.
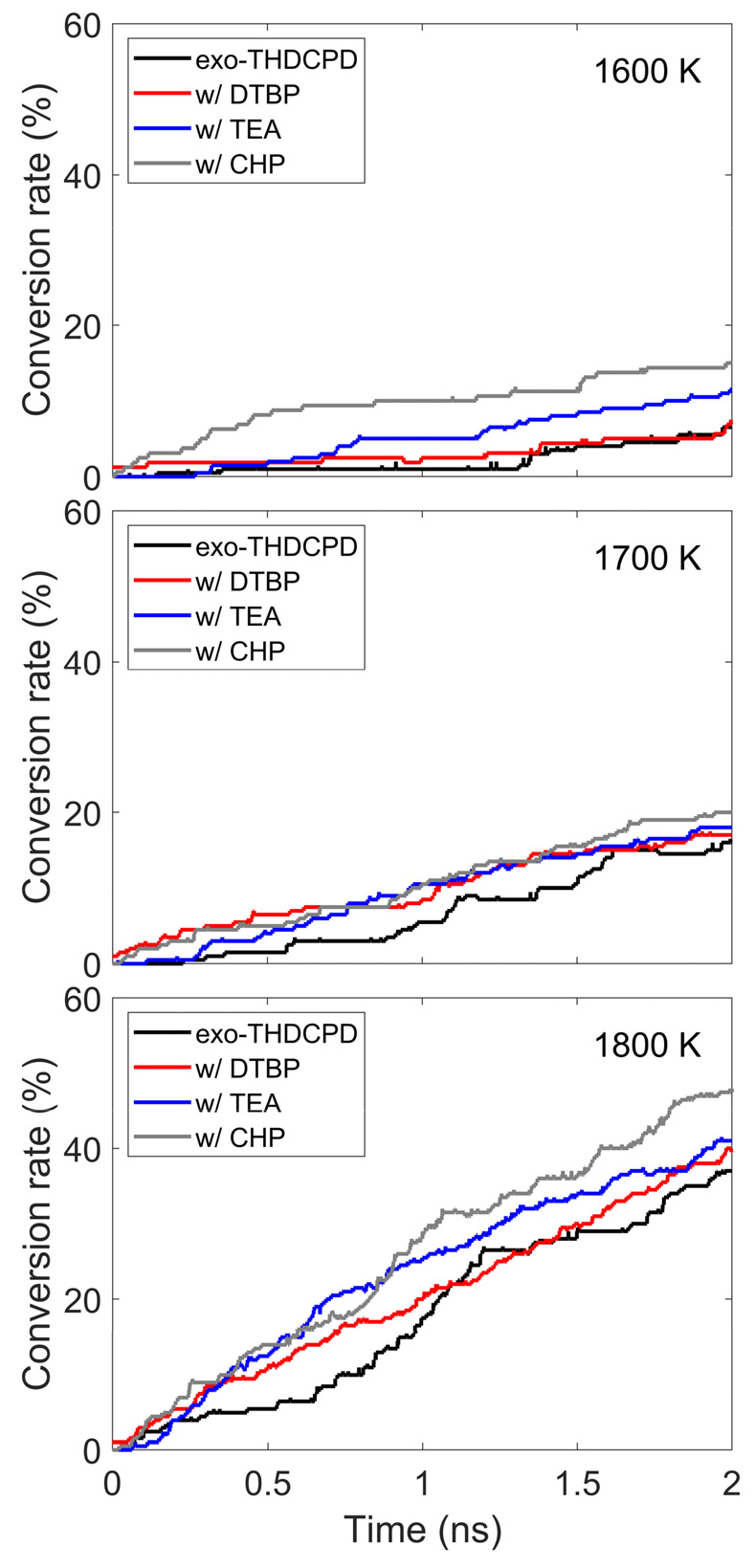
Fig. 3는 온도의 역수에 따른 각 개시제 종류별 반응 속도 상수(k)를 나타낸다. 이를 아레니우스식에 적용하여 총괄 반응의 활성화 에너지(Ea) 및 빈도인자(pre-exponential factor)를 계산하였다. CHP가 첨가된 경우, 1600 K에서 반응 속도 상수는 순수 연료보다 최대 5.6% 증가하였다. TEA를 포함할 경우, 1600 K에서 3.6% 증가했으나, 1800 K에서는 순수 연료와 거의 유사한 반응 속도를 보였다. DTBP를 첨가했을 때는 온도에 따른 반응 상수가 순수 연료와 거의 유사함을 확인하였다.
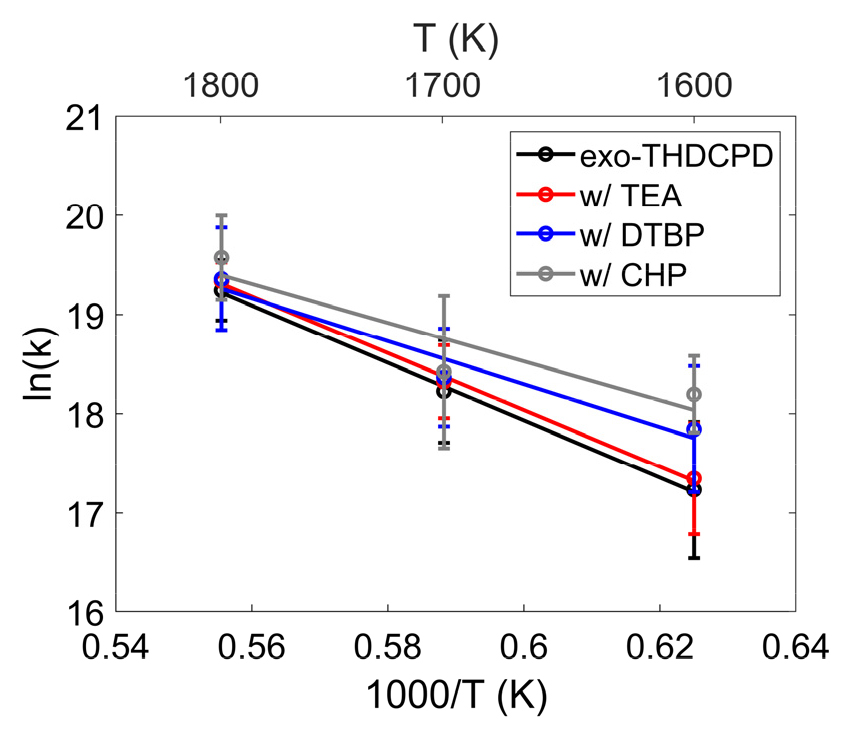
화학 개시제 종류에 따른 활성화 에너지, 빈도인자, 그리고 순수 연료 대비 활성화 에너지의 증감률을 비교하여 Table 3에 정리하였다. 먼저, 순수 연료의 실험 결과[22]와 본 연구에서 얻은 활성화 에너지는 잘 일치했으나, 빈도인자에서는 차이를 보였다. 실험과 시뮬레이션에서 적용된 온도가 크게 다르기 때문에 이러한 차이가 발생하였다. 활성화 에너지는 전환율이 높을수록 감소하는 경향을 보였다. CHP를 혼합하면 순수 연료 대비 활성화 에너지가 약 32.44% 감소하며, TEA를 첨가한 경우에도 25.18% 감소하였다. 따라서 CHP와 TEA가 비교적 효과적인 연료 첨가제 역할을 하는 것으로 확인되었다. 이에 반해, DTBP를 첨가했을 때는 활성화 에너지가 1.11% 감소하여 exo-THDCPD 연료의 열분해에 미치는 영향이 미미한 것으로 나타났다.
Table 3.
Comparison of activation energy and pre-exponential factor with different initiators. Uncertainties were obtained from ensemble-averaged simulations with a 95% confidence level
| Case | Ea(kcal/mol) | Log A | Percent change(%) |
| Neat fuel[22] | 62.40 | 13.76 | - |
| Neat fuel | 57.58 ± 1.61 | 15.34 | - |
| w/ DTBP | 56.94 ± 1.18 | 15.30 | ↓1.11 |
| w/ TEA | 43.08 ± 1.28 | 13.60 | ↓25.18 |
| w/ CHP | 38.90 ± 1.10 | 13.15 | ↓32.44 |
3.2 주요 생성물 선택도에 대한 개시제의 영향
화학 개시제는 라디칼 생성 유도, 반응 속도 향상, 생성물 선택도 조절, 그리고 저온에서의 높은 반응성 등 여러 측면에서 중요한 역할을 한다[23]. 따라서 본 연구에서는 열분해 주요 생성물인 C1-C4 파라핀 및 C2-C4 올레핀의 선택도를 분석하였다.
Fig. 4는 개시제 종류에 따른 C1-C4 파라핀 생성물의 선택도를 시간에 따라 보여준다. 순수 연료는 모든 온도에서 C1-C4 파라핀 생성물에 대한 낮은 선택도를 보였다. 이와 달리, TEA를 첨가한 조건에서는 C1-C4 파라핀의 선택도가 가장 높았다. 이는 TEA가 분해되면서 생성된 CH3 라디칼이 연료와 수소 추출 반응을 통해 CH4로 전환되는 반응이 활성화되었기 때문이다. 특히, 1800 K에서는 TEA가 분해되면서 생성된 CH3와 C2H5 라디칼이 연료와 추가 반응하면서 CH4 및 C2H6가 생성되어 반응 초기에 선택도가 급격히 증가했다. 그러나 시간이 지남에 따라 다른 화학종이 생성되면서 C1-C4 파라핀의 선택도는 점차감소하는 경향을 보였다. DTBP 또한 TEA와 마찬가지로 수소 추출 반응을 통해 C1-C4 파라핀의 선택도를 증가시켰다. 반면, CHP를 첨가한 경우 순수 연료와 거의 차이가 없었다.
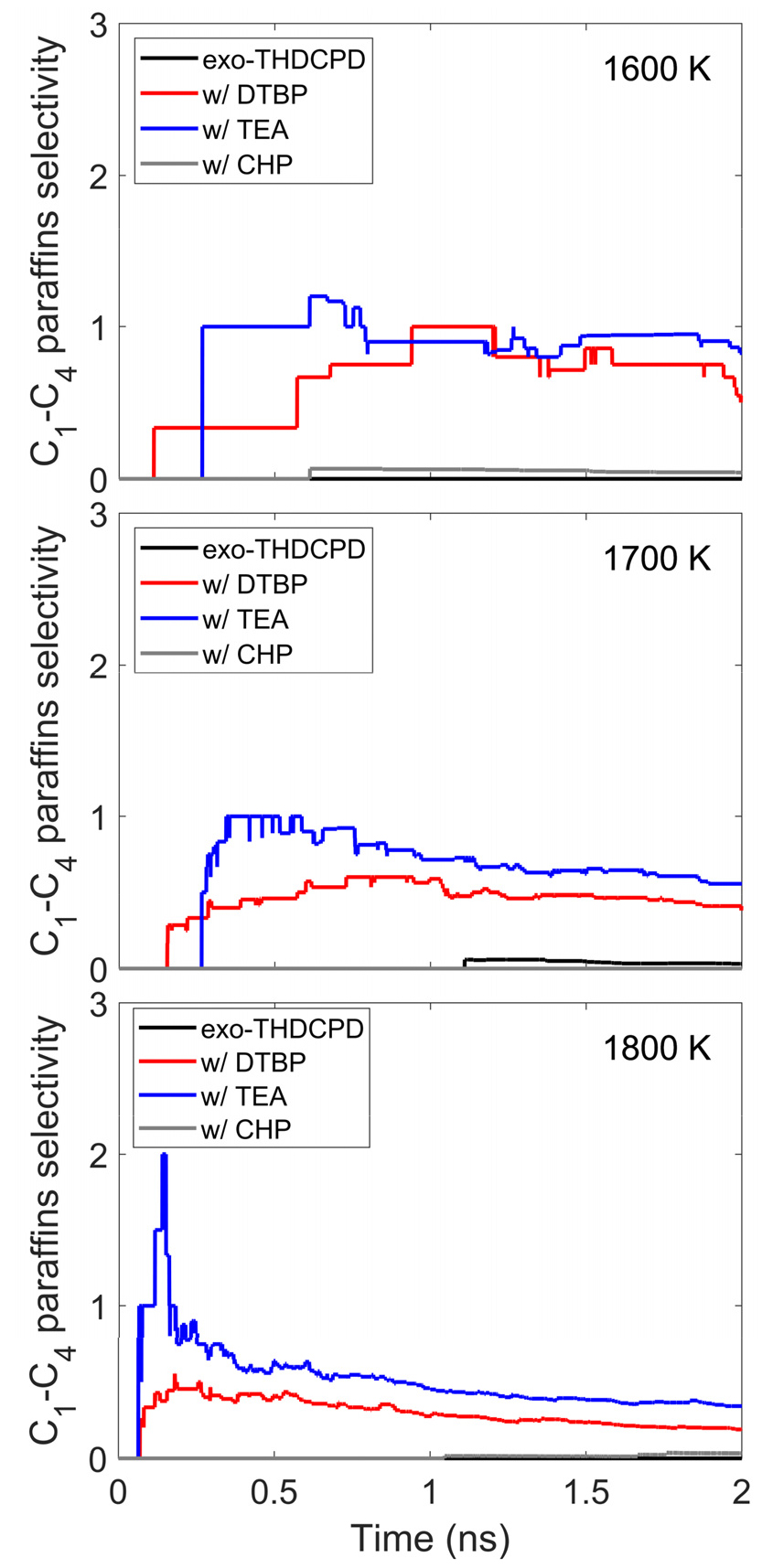
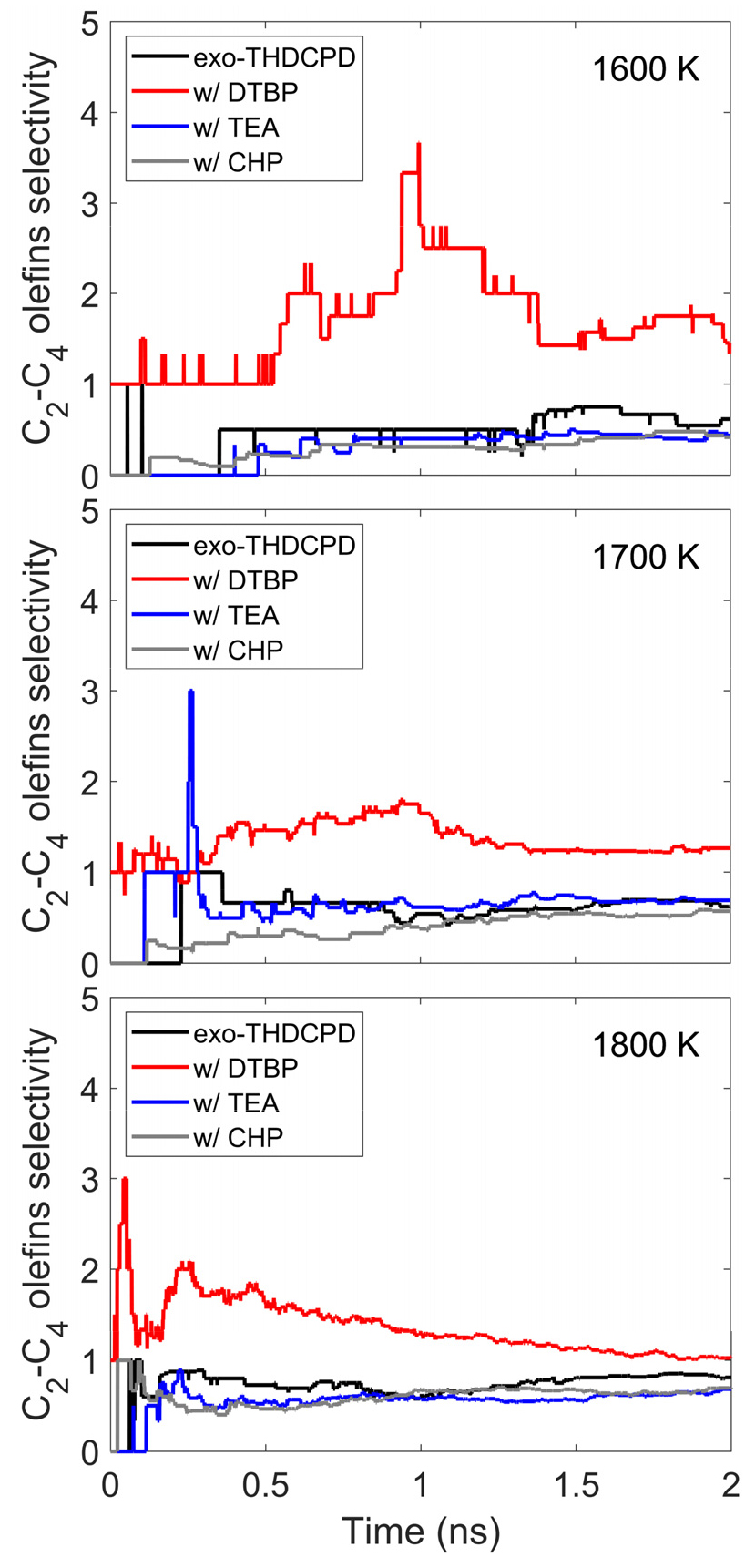
Fig. 5는 온도와 개시제 종류에 따른 C2-C4 올레핀 생성물의 선택도 변화를 나타낸다. 모든 조건에서 C2-C4 올레핀의 주요 생성물은 C2H4로 확인되었다. DTBP를 사용한 경우, C2-C4 올레핀 선택도가 가장 높게 나타났다. 또한, 반응 시작 시점부터 C2-C4 올레핀 생성이 관찰된다. 이는 초기 1500 K 열평형 시뮬레이션에서 DTBP 분해로 인해 생성된 C3H6와 C4H8 같은 올레핀 생성물 때문이다. 하지만, TEA와 CHP가 포함된 경우에는 순수 연료와 비교했을 때 C2-C4 올레핀 선택도에 큰 변화가 나타나지 않았다.
3.3 PAH 전구체 생성에 대한 개시제의 영향
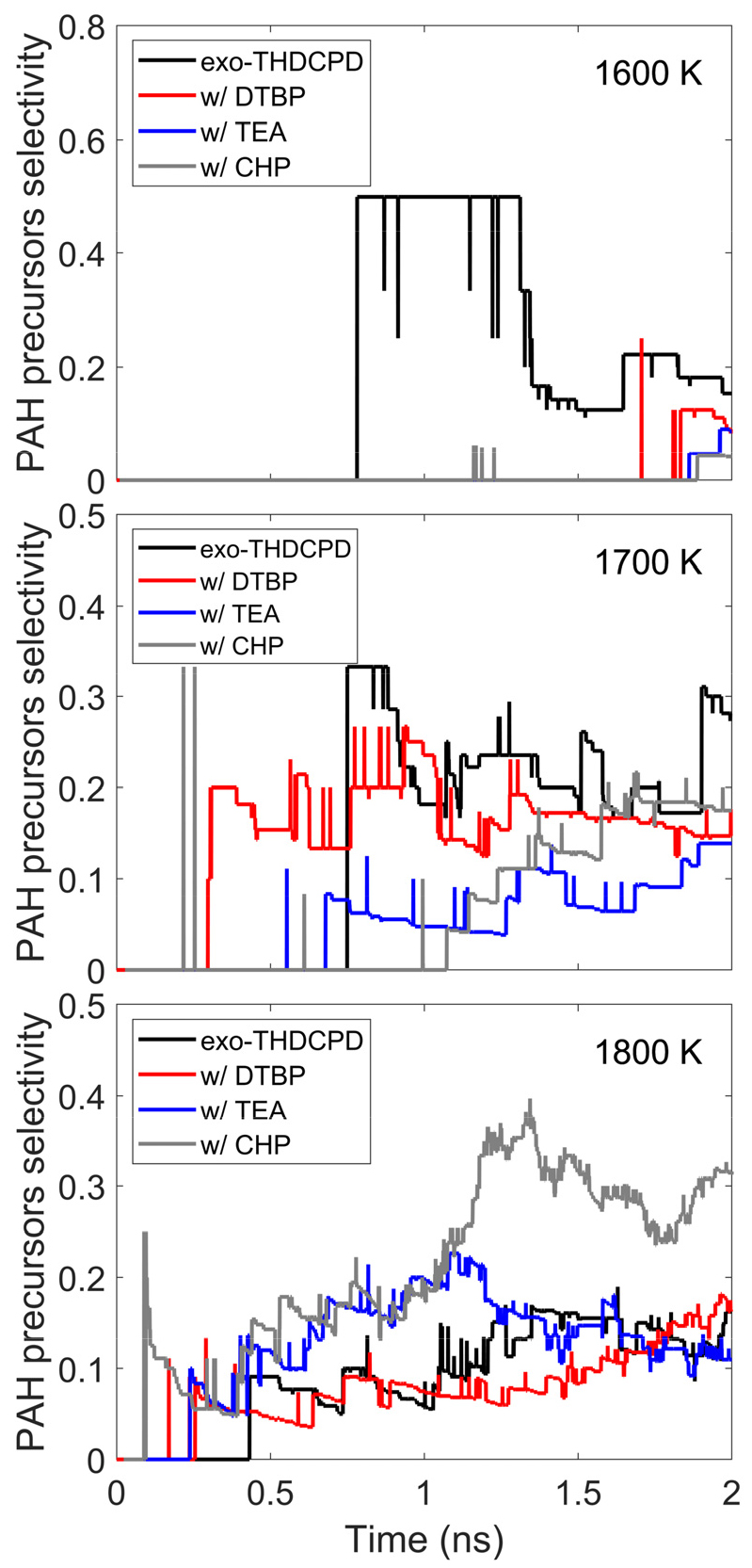
다환방향족탄화수소(PAHs)는 연료의 열분해 과정에서 생성되는 화학종이다. PAH는 주로 저분자량의 불포화 탄화수소 전구체의 축합 반응을 통해 생성된다. 이러한 과정은 코크 생성의 초기 단계를 이룬다. 따라서 PAH 전구체 분석을 통해 연료의 열분해 과정에서의 코크 생성을 예측할 수 있다[24]. 특히, C2H2와 C4H6 화합물은 PAH 형성의 초기 단계와 Diels-Alder 반응을 거쳐 코크 생성에 중요한 역할을 한다[25]. 본 연구에서는 C2H2, C3H3, C4H5, C4H6, C5H6와 C6H6를 PAH 전구체로 정의하였고, 개시제가 이러한 전구체의 생성에 미치는 영향을 평가하였다[26].
Fig. 6는 개시제에 따른 PAH 전구체의 선택도를 비교한 결과이다. 1600 K에서 순수 연료는 전환율이 낮음에도 불구하고 PAH 전구체의 선택도가 매우 높게 나타났다. 이는 순수 연료의 열분해 과정에서 C4H6의 생성률이 상대적으로 높았기 때문이다. 반면, 개시제를 적용하면 전환율은 증가하지만, PAH 전구체의 선택도는 오히려 낮아지는 경향을 보인다. 예를 들어, 1700 K 조건에서는 개시제의 영향으로 PAH 전구체의 생성 시점은 순수 연료보다 빨라졌다. 하지만, 약 0.7 ns 이후 선택도는 순수 연료보다 낮게 유지되었다. 1800 K에서는 PAH 전구체가 더욱 빠르게 생성되었다. DTBP는 순수 연료와 유사한 경향을 보였다. TEA는 PAH 전구체의 선택도가 상승하여 순수 연료보다 높은 선택도를 나타낸 후, 반응 후반부에 유사한 수준으로 다시 감소하는 경향을 보였다. 이와 달리, CHP가 포함된 조건에서는 1 ns 이후 PAH 전구체의 선택도가 급격히 증가하였다. 이러한 이유는 CHP의 열분해 과정에서 생성된 라디칼이 PAH 전구체로 빠르게 전환되었기 때문이다. 개시제가 생성물 선택도에 미치는 영향은 반응 경로 분석을 통해 다음 장에서 구체적으로 논의된다.
3.4 개시제 종류에 따른 연료의 반응 경로 메커니즘 분석
본 연구에서는 분자동역학 시뮬레이션을 통해 예측된 순수 연료와 개시제를 첨가한 연료의 분해 반응 경로를 비교 분석하였다.
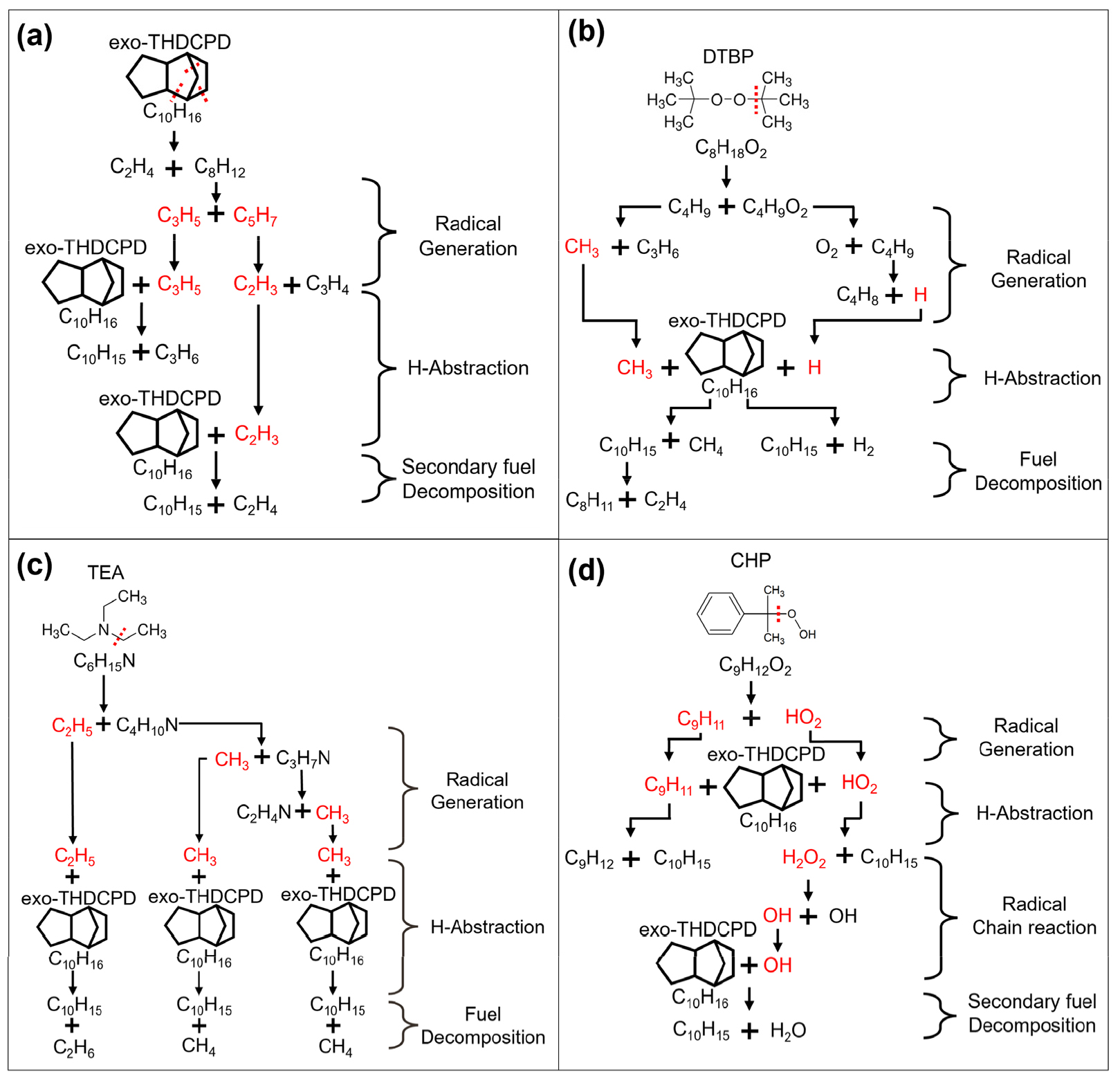
우선, 반응 경로 분석을 통해 순수 연료의 열분해 반응 메커니즘을 Fig. 7(a)에 나타냈다. 그림에서처럼, 순수 연료는 초기 단계에서 C-C 결합이 해리되면서 C2H4와 C8H12로 분해된다. 이후, 분자량이 큰 C8H12는 C3H5와 C5H7의 라디칼로 2차 분해되며, 생성된 C5H7은 다시 C2H3와 C3H4로 3차 분해된다. 이러한 분해 과정에서 생성된 라디칼은 수소 추출 반응을 통해 연료의 2차 열분해를 유도한다. 이전 연구에서도 본 연구와 유사한 반응 경로가 예측된 바 있다[27].
Fig. 7(b)는 DTBP가 포함된 연료 혼합물의 분해 반응 경로를 보여준다. 앞서 언급했듯이, DTBP는 반응 초기에 C-O 결합의 해리로 인해 빠르게 C4H9과 C4H9O2로 분해된다. 또한, C4H9O2는 C4H9과 O2로 2차 분해 과정을 거친다. 그림에서 보듯이, 이렇게 생성된 C4H9 라디칼은 C3H6 및 C4H8과 같은 올레핀 생성물로 추가 분해된다. 이는 Fig. 5에서 확인한 C2-C4 올레핀에 대한 높은 선택도가 DTBP의 분해 과정에서 생성된 올레핀 화합물 때문임을 시사한다. 앞서 생성된 라디칼들은 연료의 2차 분해 반응에 참여하여 CH4를 생성한다. 이로 인해 DTBP가 높은 C1-C4 파라핀 선택도를 나타냈다.
TEA가 혼합된 연료의 반응 경로를 Fig. 7(c)에 제시하였다. TEA는 먼저 C-N 결합이 해리되면서 CH3와 C2H5 라디칼로 분해된다. 앞선 과정과 유사하게, 생성된 라디칼들은 수소 추출 반응 통해 연료를 추가 분해시킨다. 이 과정에서 CH4 및 C2H6와 같은 파라핀 화합물이 생성된다. 이는 TEA 주입이 C1-C4 파라핀 선택도를 증가시키는 메커니즘으로 작용했음을 의미한다.
끝으로 Fig. 7(d)에서는 CHP 개시제가 연료의 분해 반응 경로에 미치는 영향을 분석하고자 한다. 다른 개시제와 마찬가지로, CHP는 반응 초기에 HO2와 C9H11과 같은 라디칼로 분해된다. 그림에서처럼, C9H11 라디칼은 연료의 분해 반응에 기여한다. 흥미롭게도, HO2는 연료와 반응하여 H2O2로 전환되고, 이후 OH 라디칼로 다시 해리된다. 생성된 OH 라디칼은 연료 분자와 빠르게 연쇄 반응한다. 이러한 라디칼 연쇄 반응이 CHP를 첨가한 혼합 연료에서 전환율이 가장 높게 나타난 주요 원인이다.
4. 결 론
본 연구에서는 ReaxFF 분자동역학 시뮬레이션을 활용하여 exo-THDCPD의 분해 반응을 분석하였다. 특히, DTBP, TEA, CHP와 같은 화학 개시제 첨가가 연료의 분해 생성물, 반응 동역학 및 반응 메커니즘에 미치는 영향을 조사하였다. 이를 통해 다음과 같은 결론을 도출하였다.
개시제 종류에 따라 연료의 전환율이 크게 변화했다. 특히, CHP를 주입한 경우 가장 높은 전환율을 보였으며, 활성화 에너지를 약 32% 감소시켰다. TEA 또한 유사한 효과를 나타냈다. 반면, DTBP는 전환율과 활성화 에너지의 변화가 거의 없었다.
개시제 종류에 따라 생성물 분포도에 차이가 확인되었다. DTBP는 C1-C4 파라핀 및 C2-C4 올레핀의 선택도를 증가시켰고, TEA는 CH4와 C2H6 생성을 향상시켰다. 이와 달리, CHP는 주요 생성물 분포 변화에 미치는 영향이 상대적으로 적었다.
PAH 전구체 선택도 분석을 통해, 높은 전환율을 보인 TEA가 PAH 전구체의 생성을 억제함을 확인했다. CHP는 1800 K에서 PAH 전구체의 선택도를 크게 증가시켰다.
반응 경로 분석 결과, 개시제는 C-N 및 C-O 결합의 해리를 통해 라디칼로 분해된다. 특히, TEA와 CHP의 분해로 생성된 라디칼이 수소 추출 반응을 유도하여 연료의 분해율을 증가시켰다.
이러한 결과를 통해 개시제 선정이 연료의 전환율, 생성물 선택도, 코크 생성률 및 반응 메커니즘에 미치는 영향을 확인하였다. 본 연구 결과를 바탕으로 코크 생성을 최소화하고 분해 효율을 향상시킬 수 있는 개시제 최적화 연구가 진행 중이다.
