

1. 서 론
2. 연구 방법
2.1 화학 반응 인공신경망
2.2 학습 조건 및 결과
3. 결과 및 논의
3.1 총괄 반응식
3.2 온도 및 화학종 농도 예측
3.3 점화 지연 시간 예측
4. 결 론
1. 서 론
전 세계적으로 화석 연료의 사용이 지속 되어 오면서, 이산화탄소 증가에 의한 지구 온난화 및 기후변화 위기가 큰 문제로 대두되었다. 이를 해결하기 위해서는 발전 방식의 변화를 통한 탄소 중립 실현이 필요하므로 지속 가능한 발전을 위한 신재생에너지 사용 연구가 활발하게 진행되고 있다. 하지만, 현재의 기반 시설 및 기술을 통해서는 에너지 수요의 전체 양을 신재생에너지로 대체하는 데 한계가 있다[1]. 또한, 현재의 신재생에너지기술의 간헐성에 대응하는 것이 필요하다. 따라서, 수요에 맞는 에너지를 공급하면서도, 이산화탄소 배출량을 감소할 수 있는 무 탄소 연료의 사용이 제안되었다[2].
이 중, 수소는 연료 전지 및 연소 시스템을 통한 친환경적 에너지 생산이 가능하다는 장점이 있다. 하지만, 수소는 저장과 운송이 쉽지 않기 때문에, 이를 보완하기 위해 암모니아의 활용에 관한 관심이 높아졌다[2]. 암모니아는 수소 에너지 캐리어로써의 활용 가능성이 크며, 고체 산화물 연료 전지의 연료로 다양하게 연구가 진행됐다. 암모니아의 높은 에너지 밀도라는 장점뿐만 아니라, 100년이 넘는 기간 동안 산업현장에서 다양하게 사용되어왔다는 점 덕분에, 관련 기반 시설이 이미 마련되어 있다는 장점도 가진다. 따라서, 암모니아를 활용한 지속할 수 있고 친환경적인 에너지 생산이 주목받고 있다.
탄소 중립 목표 달성을 위한 저탄소 및 무 탄소 에너지 기술 발전 중 하나로, 암모니아 연소기술이 현재까지 다양하게 연구되었다[3]. 암모니아의 혼합연소 및 직접 연소기술의 개발을 위해서는 연소 반응에 대한 이해를 바탕으로 효율 증대와 질소 산화물 생성 최소화를 위한 노력이 필요하다. 실험적 연구와 더불어 전산 유체역학 시뮬레이션을 이용하면 이를 수치적으로 모사하여 연소 생성물에 대한 예측을 통한 연소 환경 최적화를 구현할 수 있다. 이러한 시뮬레이션 계산을 위해서, 암모니아의 화학 반응 메커니즘에 대한 확립이 필수적이다[4].
암모니아 연소 연구에서 널리 사용되는 전체 메커니즘은 [5,6,7] 등이 있으며, 전체 메커니즘은 반응식의 및 화학의 개수가 많다는 특징이 있다. 이러한 특징 때문에, 2차원 이상의 시뮬레이션에서는, 전체 반응식 사용 시, 계산 비용이 매우 커진다는 한계가 있다[8]. 이러한 한계 극복을 위하여, 전체 반응식에서 반응식의 개수를 줄여 축소 반응식을 개발하는 연구가 활발히 진행됐다. 예를 들어, [9]의 축소 반응식은 총 22개의 종과 92개의 반응식으로 구성된다. 하지만, 축소 반응식 또한, 고차원 시뮬레이션 계산에서는 계산 시간이 오래 소요될 수 있으므로, 더욱 획기적인 계산량 감소가 요구되고 있다. 이를 보완하기 위한 것이 총괄 반응식으로, 중간 생성물 등을 생략하여 더 적은 수의 종과 반응식으로 구성되면서도, 생성물 농도를 예측하고, 층류 연소 속도 및 온도 등을 실제 실험 결과와 유사하게 모사하는 것을 목표로 한다. 이처럼 총괄 반응식 사용 시, 암모니아 연소 연구의 계산 접근성과 효율성을 높일 수 있다. 현재까지 다양한 문헌에서 총괄 반응식 개발 연구를 진행하였다. 특히, 탄화수소 연료의 연소 과정 묘사를 위한 총괄 반응식은 오랜 시간에 걸쳐 연구와 검증이 진행되었다.
또한, 다양한 연구에서 연소 반응을 모사의 정확도를 높이거나, 계산 효율성을 개선하기 위하여 기계학습을 활용하였다[10,11,13]. [12]는 직접 수치 모사 기법(Direct numerical simulation)에서의 비예혼합 난류화염과 냉각된 벽의 상호작용 계산 비용의 감소를 위하여 인공신경망(Articifial neural network)을 사용하였고, 높은 수준의 예측 정확도를 보이면서도 총괄 반응식에 비하여 CPU 계산 비용을 25배 감소시켰다. 또한 [13]은 암모니아 연소의 화학 반응식의 최적화를 딥러닝 모델을 활용하였고, 높은 압력 조건에서 점화 지연 시간에 대한 성공적 예측을 구현하였다.
따라서, 본 연구는 암모니아 연소 과정의 전산 유체역학 시뮬레이션 계산 비용 감소를 위한 총괄 반응식의 개발을 목표로 한다. 이를 위하여, 본 연구에서는 기계학습 방법 중 화학 반응 인공신경망을 활용하여 기존의 전체 반응식 결과를 모사하였고, 온도 및 화학종 농도 예측과 점화 지연 시간 예측의 성능 평가를 진행하였다.
2. 연구 방법
2.1 화학 반응 인공신경망
2.1.1 계산 원리
본 연구에서는 화학 반응 인공신경망(Chemical reaction neural network, CRNN)을 활용하여, 총괄 반응식을 개발하였다. 화학 반응 인공신경망은 입력층과 출력층 사이에 하나의 은닉층이 포함된 구조를 가지며, [14,15,16]에서 제안된 방법이다. 이 인공신경망은 화학종과 반응식의 개수 지정 후, 훈련하여 화학 반응식을 완성한다. 인공신경망에서는 데이터가 입력층, 은닉층, 그리고 출력층을 거치면서, 가중치와 활성 함수에 의해 계산이 되어 처음의 값과 비선형적 관계의 출력값을 만들어낸다. 이때, 손실 함숫값을 최소화하는 방향으로 훈련이 진행되면서 가중치의 값이 갱신된다. 전통적인 딥러닝을 이용하면 여러 개의 은닉층을 거치면서 가중치의 값이 최적화되어 원하는 출력을 얻을 수 있었지만, 훈련 결과를 설명할 수 없다는 특징 때문에 이를 블랙박스(Black box)라고 불렀다. 화학 반응 인공신경망은 이러한 한계를 극복하며 딥러닝 구조 자체에 설명 가능성을 부여한 방법이라고 할 수 있다. 화학 반응 인공신경망에서는 가중치 자체가 화학 반응식의 물리적 정보를 설명한다. 화학 반응 인공신경망의 원리는 다음과 같다.
A, B, C, D의 네 가지 화학종으로 구성된 하나의 반응식을 식 (1)이라고 한다면, 이 반응식의 반응속도는 식 (2a)와 같이 계산된다. 식 (2a)를 지수함수로 변형하여 나타내면 식 (2b)가 되는데, 이 식의 계산이 화학 반응 인공신경망의 계산 구조로 모사 된다. 이때, 각 화학종의 농도의 로그 값이 인공신경망의 입력층 데이터를 나타내며, 이 값에 곱해진 반응 차수가 입력층과 은닉층 사이의 가중치 값으로 대표된다. 식 (2)를 통해서 계산되는 출력값은 생성물의 생성률이다. 즉, 출력층의 노드 개수는 개발하려는 반응식에 포함된 화학종의 개수와 같다. 아레니우스식의 활성화 에너지인 와 충돌 빈도 인자 A, 그리고 온도 정비 인자 b 또한 화학 반응 인공신경망을 통해 최적화하였다. 화학 반응 속도상수 k를 계산하는 아레니우스식은 식 (3a)와 같다. 이 식을 식 (3b)와 같이 변형시키고, 입력층에 와 값을 갖는 노드를 추가시켜 아레니우스식을 화학 반응 인공신경망 구조에 포함할 수 있다.
2.1.2 적용 방법
본 연구에서 암모니아 연소를 위해 사용한 화학 반응 인공신경망의 구조는 Fig. 1(a)과 같다. 입력층에서는 총괄 반응식에 포함할 화학종의 시간마다 농도와 온도에 대한 정보를 받는다. 입력층에서 은닉층으로 진행될 때 결정되는 입력층 가중치는 반응 차수를 나타낸다. 하나의 층으로 구성된 은닉층에 포함되는 노드의 개수는 총괄 반응식에 포함되는 반응식의 개수를 나타낸다. 은닉층을 지나 출력층으로 진행될 때 결정되는 출력층 가중치는 화학량론 계수(stoichiometric coefficient)를 나타낸다. 입력층의 편향(bias)을 이용하여 아레니우스식의 상숫값을 결정하게 된다.
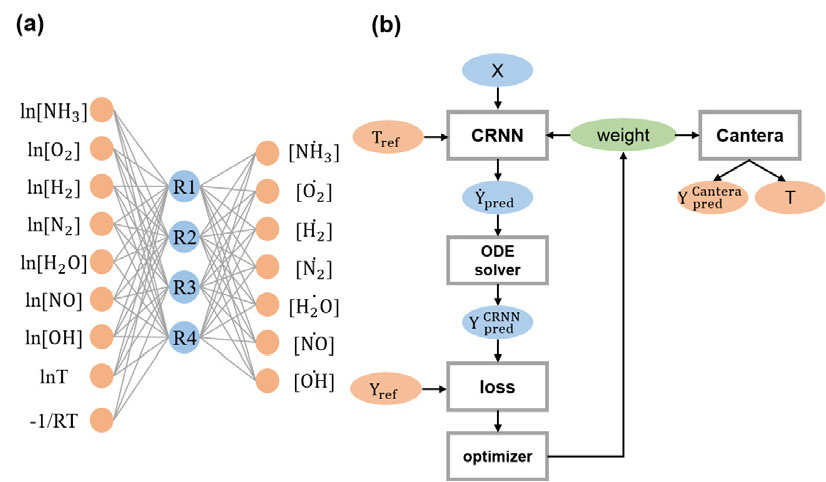
이렇게 구성된 화학 반응 인공신경망의 계산 과정을 정리해보면 식 (4)와 같다. 이 과정에서 미분방정식을 풀기 위해 [14]에서 제시된 것과 같이 Julia를 이용하여 훈련을 진행하였다. 손실함수는 와 의 평균 절대오차를 사용하다.
본 연구에서의 화학 반응 인공신경망 학습 및 성능 평가를 위한 계산 흐름도는 Fig. 1(b)과 같다. 화학 반응 인공신경망을 통해 결정된 총괄 반응식의 성능 평가를 위해 Cantera를 이용하였다. 0 차원 반응기의 암모니아 연소 생성물 농도 및 온도는 Cantera을 통해 계산되어 전체 반응식을 이용하여 계산한 기준 데이터와 비교하였다. 연소의 점화 지연 시간에 대해서도 성능 평가를 진행하였다.
2.2 학습 조건 및 결과
2.2.1 학습 데이터
기존 암모니아 연소를 모사하는 전체 반응식 중, Okafor [5] 반응식을 본 연구의 훈련 데이터 생성에 사용하였다. 본 연구의 총괄 반응식은 총 4개의 식을 포함한다. 총괄 반응식에서는 각 단계에서의 중간 생성물을 생략하고, 전체적인 결과를 모사하는 것이 목표이기 때문에, 포함되는 화학종의 종류를 적절하게 결정하는 것이 필요하다. 따라서, 본 연구에서는 총 7개의 화학종 을 선택하였다. 포함되는 화학종이 평형상태에서의 전체 몰분율의 대부분을 대표할 수 있을 때, 총괄 반응식을 이용한 온도의 예측 정확도가 올라갈 수 있다. 평형상태에서 선택된 7개의 화학종이 차지하는 비율을 각각의 온도와 당량비별로 나타내면 Fig. 2와 같다. 고온에서 그 비율이 감소하지만, 2400 K에서도 93% 이상을 차지하는 것을 확인할 수 있다. 90% 이상의 평형상태 몰분율을 포함하기 위하여 화학종은 7개로 결정하였다. 이때, 데이터 세트 계산을 위한 온도, 당량 비, 시간 간격, 종료 시각의 초기 조건은 Table 1에 나타나 있다. 훈련과 검증 데이터는 초기 온도 조건을 기준으로 구분하였으며, 훈련 데이터는 1460, 1480, 1500, …, 1720, 1740 K의 조건, 검증 데이터는 1470, 1490, 1510, …, 1730, 1750 K 의 조건을 적용하였다.
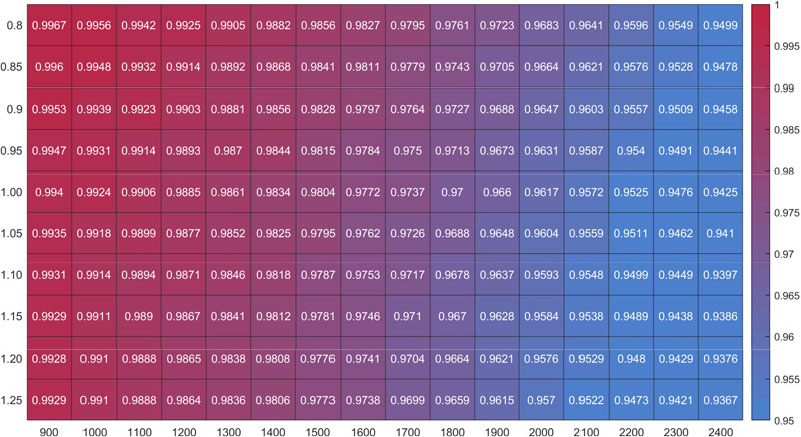
Table 1.
Information of data set for training CRNN
2.2.2 학습 결과
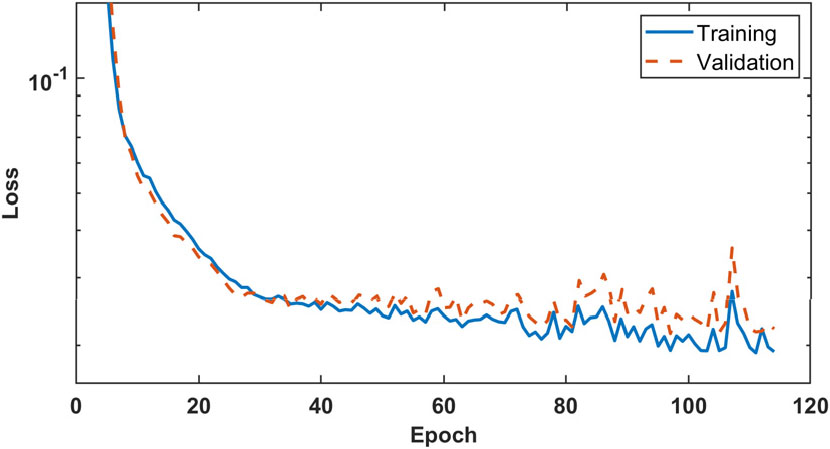
이를 통해 총 7개의 화학종을 반응식에 포함하도록 하는 화학 반응 인공신경망의 구조를 Fig. 1(a)과 같이 설정하여 학습을 진행하였다. 학습 진행에서의 손실함수 값은 Fig. 3와 같다. 계산은 약 100 에포크까지 진행되었으며, 손실함수의 값이 이후 수렴하는 것을 확인할 수 있다.
3. 결과 및 논의
3.1 총괄 반응식
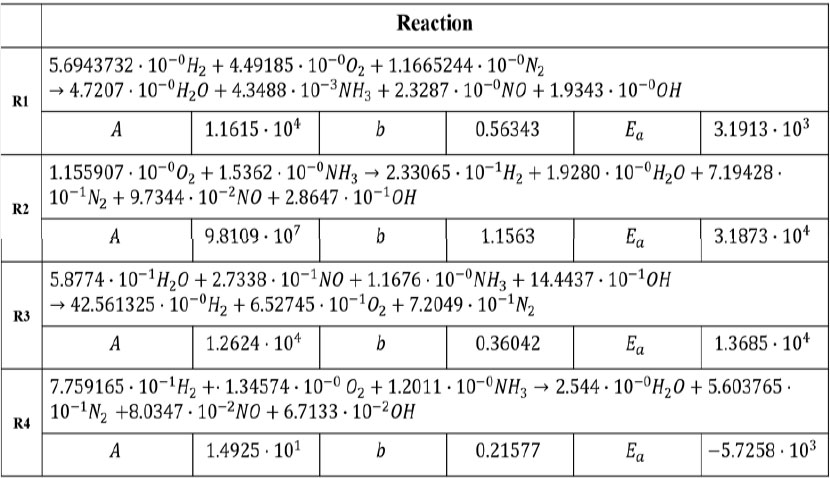
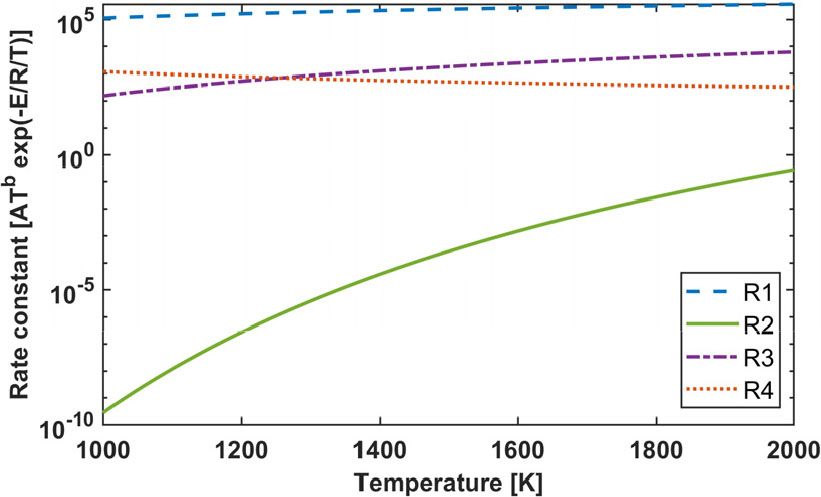
학습을 통해 얻은 4개의 반응식과 7개의 화학종이 포함된 총괄 반응식은 Fig. 4와 같다. 각각의 반응식의 화학량론 계수가 결정되었으며, 아레니우스식의 상수 또한 결정되었다. 두 번째 반응식이 산소와 암모니아를 반응물로 하는 반응식을 대표한다. 생성된 물과 질소 산화물, 암모니아 및 수산화물의 반응이 세 번째 반응식으로 나타나며, 수소, 산소, 질소를 반응물로 하는 첫 번째 반응식, 그리고 수소, 산소, 암모니아를 반응물로 하는 네 번째 반응식을 확인할 수 있다. 즉, 4단계에 걸쳐 암모니아 연소 반응을 모사하기 때문에, 두 번째 반응식에서의 생성물이 첫 번째, 세 번째, 네 번째 반응식을 계속해서 거치며 추가 반응이 진행되는 것을 확인할 수 있다. Fig. 5는 4개의 반응식의 반응속도 상수를 나타낸 것이다. 온도가 변함에 따라 두 번째 반응식의 반응속도상수 값이 가장 크게 변하는 것을 확인할 수 있다. 반응속도 상숫값이 가장 큰 반응식은 첫 번째 반응식이었으며, 가장 작은 반응식은 두 번째 반응식이다.
3.2 온도 및 화학종 농도 예측
Fig. 6은 Fig. 4의 반응식을 이용하여 0차원 반응기에서 암모니아 연소의 시간에 따른 온도 변화 및 화학종의 각 농도 예측을 나타낸 것이다. 초기 온도 1560 K, 압력 1 atm, 당량 비 0.85의 조건에서 온도 및 농도 변화를 전체 반응식으로 계산한 경우와 본 연구에서 개발된 총괄 반응식으로 계산한 경우, 그 개형이 유사함을 확인할 수 있다. 특히, 수소 농도 예측을 살펴보면 전체 반응식으로 계산한 결과에서 점화 반응이 시작되면서 정점 점에 도달하고 다시 감소하는 개형이 확인되는데 이 경향성이 총괄 반응식에서도 구현되었고, 평형상태에 도달했을 때의 농도 예측 정확도가 높음을 확인할 수 있다. 화학종 농도 예측값뿐만 아니라, 온도의 예측 결과를 확인하여 보면, 마찬가지로 평형상태 도달 시 예측값이 전체 반응식과 본 연구에서 개발된 총괄 반응식이 서로 유사하다.
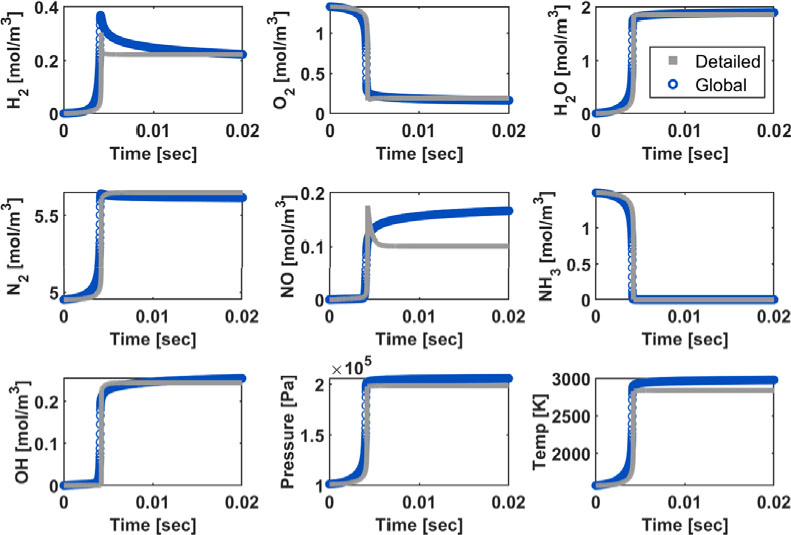
Fig. 7에서는 수소, 일산화질소 생성물 농도와 최종온도에 관한 결과 예측 정확도를 0.8에서 1.25 범위의 당량 비에서 평가하였다. 수소 농도 예측 정확도가 높은 편임을 확인할 수 있는데, 당량 비가 낮을수록 총괄 반응식이 더 정확하게 수소 농도를 예측한다. 이와 반대로, 질소 산화물의 예측은 당량 비가 낮을 때 가장 오차가 크다. 총괄 반응식과 전체 반응식의 일산화질소 농도 예측값이 차이를 보이는데, 이는 전체 반응식에서는 등의 질소 산화물이 모두 고려되지만, 본 연구의 총괄 반응식에서는 질소 산화물의 화학종 중에서는 만 포함되었기 때문이다. [17]에 의하면 총괄 반응식에서 포함되는 화학 종인 와 가 의 생성을 촉진할 수 있는데, 본 연구에서는 를 고려하지 않았으므로 해당 반응이 반영되지 않아 일산화질소 농도를 과대예측한다.
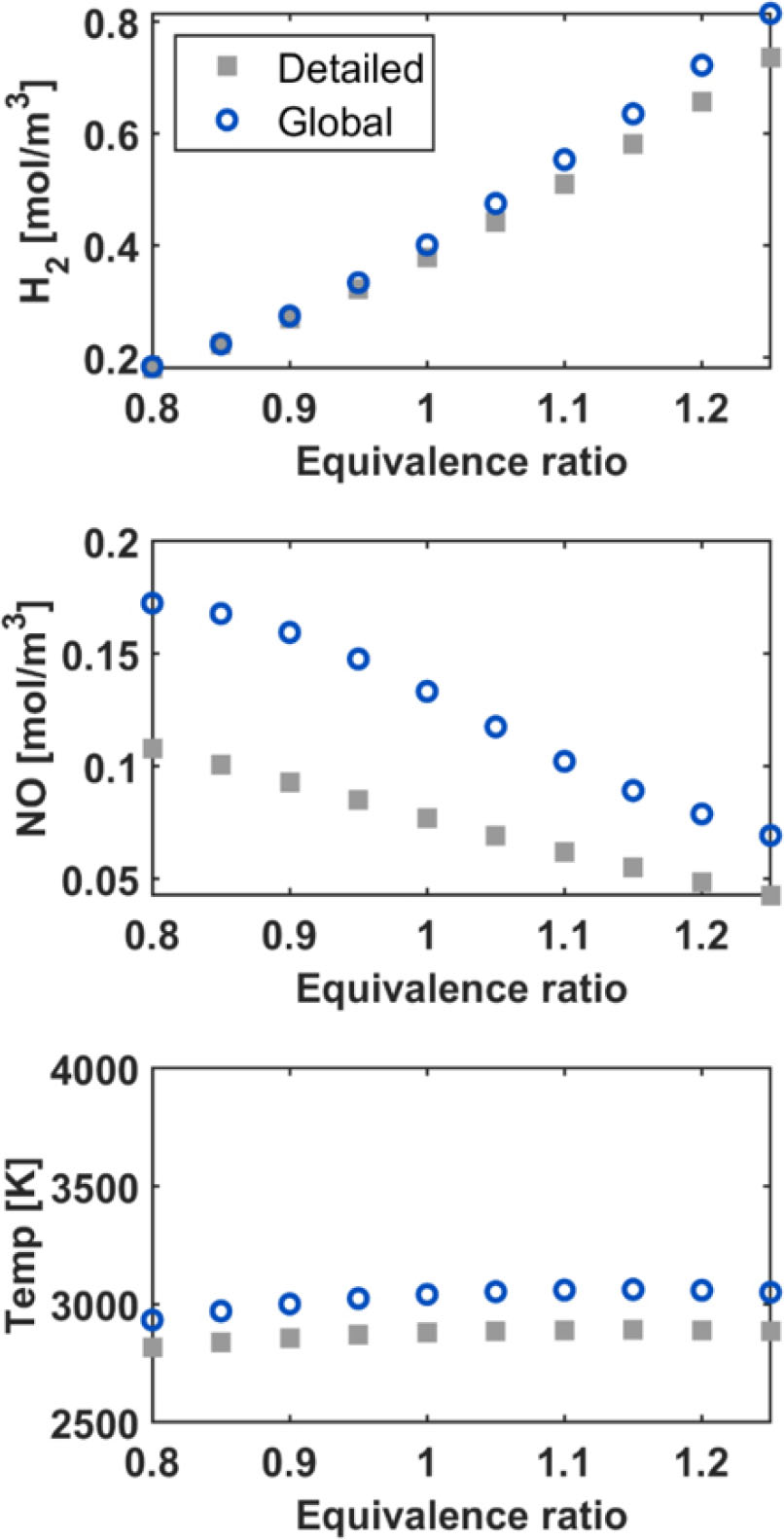
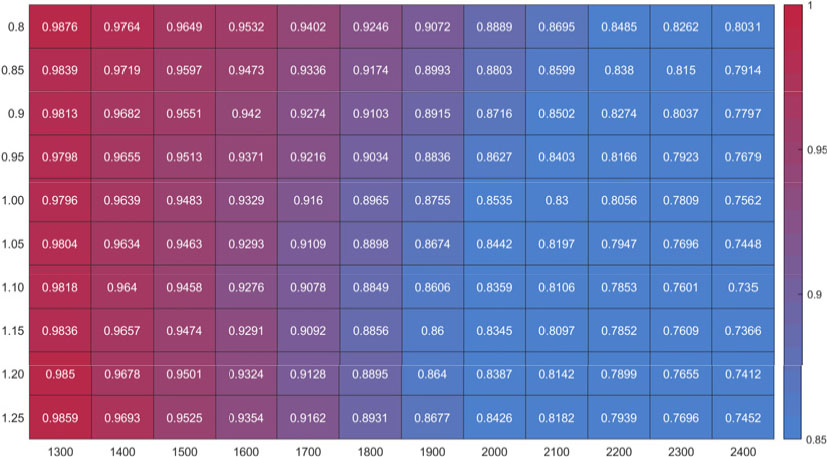
Fig. 8은 총괄 반응식과 전체 반응식의 온도 범위 1200 K에서 2400 K, 당량 비 범위 0.8에서 1.25 조건에서의 평형상태 온도 예측값을 총체적으로 비교하였다. Fig. 2에서의 온도, 당량 비에 따른 경향성이 비슷하다고 볼 수 있는데, 이는 평형상태 최종온도 예측을 위해서는 최종 생산물을 잘 대표해야 함을 알 수 있다. 이러한 이유로 인하여 본 연구의 총괄 반응식은 초기 온도가 저온일 경우 최종온도 예측값이 더 정확한 경향을 보인다. 초기 온도가 1300 K 일 때 온도예측 정확도는 약 97% 이상임을 확인할 수 있다.
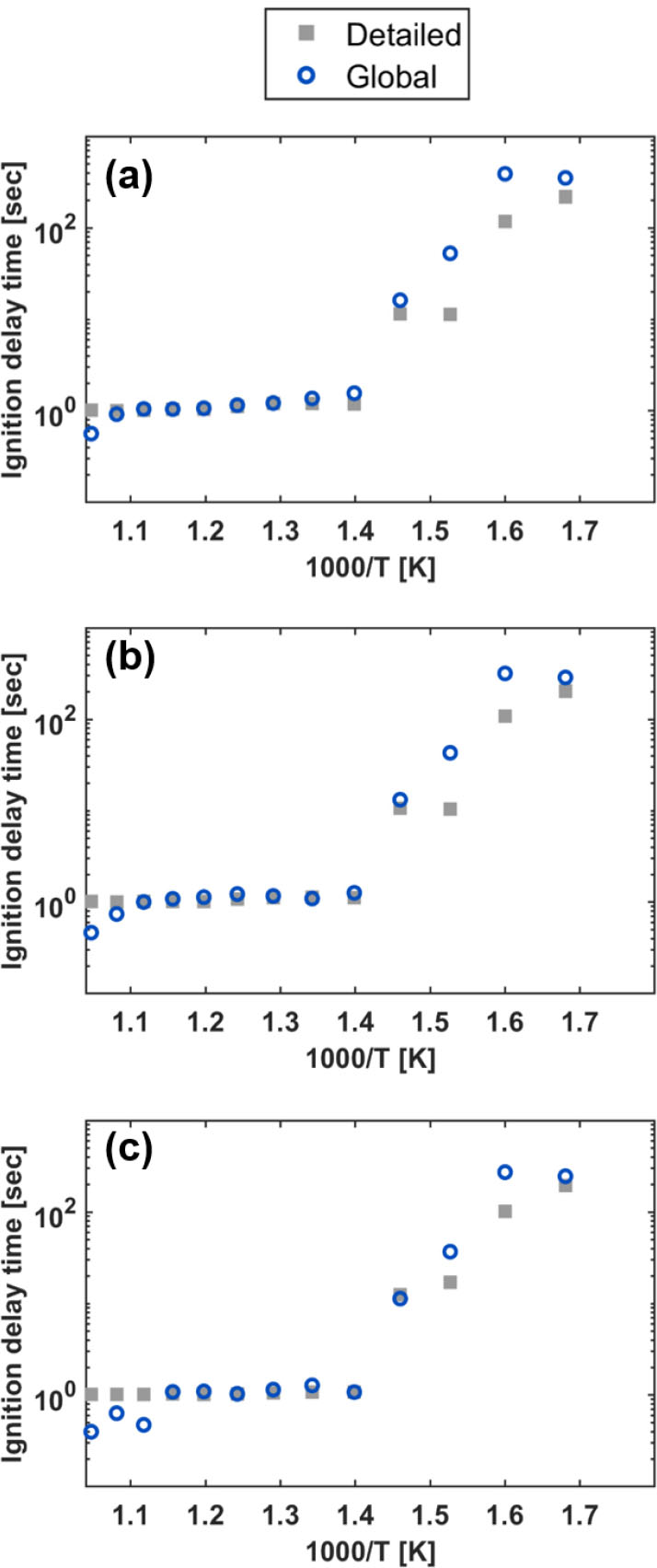
3.3 점화 지연 시간 예측
온도별, 당량 비별 점화 지연 시간 예측에 대한 성능 평가가 진행되었다. Fig. 6에서 시간에 따른 온도 및 수산화물 농도 변화 그래프에서 확인할 수 있듯이, 전체 반응식의 점화 지연 시간을 총괄 반응식이 잘 구현한다.
본 연구에서는 OH의 농도를 기준으로 점화 지연 시간을 Cantera을 이용하여 계산하였다. 차례로 당량 비 0.8 조건, 1.0 조건, 1.2 조건에서의 0차원의 반응기 점화 지연 시간 예측 결과는 Fig. 9를 통해 나타내었다. 암모니아 희박 및 과농의 넓은 범위 조건에서 개발된 총괄 반응식이 성공적으로 점화 지연 시간을 예측하는 것을 확인할 수 있다. 총괄 반응식과 전체 반응식의 점화 지연 시간 예측값 차이는 CRNN 학습에 사용한 데이터의 초기 온도 조건이 1460–1750 K의 비교적 고온 조건이라는 점에서 기인했을 것으로 추측된다.
4. 결 론
본 연구는 탄소 중립 목표 달성을 위한 저탄소 및 무 탄소 에너지 기술의 발전을 목표로, 암모니아 연소 과정의 전산 유체역학 시뮬레이션 계산 비용을 감소시키기 위해 총괄 반응식을 개발하였다. 이를 위해 화학 반응 인공신경망(CRNN)을 활용하여 기존의 전체 반응식 결과를 모사하고, 온도 및 화학종 농도 예측과 점화 지연 시간 예측의 성능을 평가하였다.
연구 결과, CRNN을 이용하여 총 4개의 반응식과 7개의 화학종을 포함하는 총괄 반응식을 개발하였다. 이 총괄 반응식은 전체 반응식과 비교하여 암모니아 연소 시의 온도 및 화학종 농도 예측에 있어 높은 정확도를 보였다. 특히, 평형상태에서의 온도예측 정확도가 97% 이상으로 나타났으며, 수소 농도 및 질소 산화물 생성 농도 예측에서도 유사한 경향성을 보였다. 또한, 점화 지연 시간 예측에서도 전체 반응식과 유사한 성능을 보이며, 다양한 조건에서 성공적으로 점화 지연 시간을 예측하였다.
본 연구에서 개발된 총괄 반응식은 기존의 전체 반응식과 비교하면 계산 비용을 크게 줄이면서도, 높은 예측 정확도를 유지한다는 장점이 있다. 이를 통해 암모니아 연소 연구의 계산 접근성과 효율성을 크게 향상할 수 있을 것으로 기대된다. 앞으로의 연구에서는 더욱 다양한 조건과 반응 환경에서 총괄 반응식의 성능을 검증하고, 이를 실용화하기 위한 추가 연구가 필요할 것이다.
따라서, 본 연구는 암모니아를 활용한 친환경 에너지 생산 기술의 발전에 이바지할 뿐만 아니라, 탄소 중립 실현을 위한 중요한 기반 기술을 제공한다는 의의가 있다.
