

1. 서 론
이산화탄소는 지구온난화 및 기후 변화에 큰 영향을 끼치는 온실가스이다. 이산화탄소의 배출량을 줄이기 위해서 탄소 포집·활용·저장(Carbon Capture Utilization and Storage) 기술들이 연구되고 있으며[1], 그 중에서도 탄화수소 건식 개질이 많은 주목을 받고 있다. 건식 개질은 탄화수소와 온실가스인 이산화탄소를 반응물로 사용하여 수소와 일산화탄소로 구성된 합성가스를 생산하는 기술이다. 합성가스는 피셔-트롭쉬 공정의 원료로 사용되어 고부가가치 물질로 전환이 가능하다[2]. 그러나, 건식 개질은 흡열 반응으로 반응에 필요한 활성화 에너지가 높다. 활성화 에너지를 낮추기 위해 촉매를 사용하기도 하지만, 촉매는 고온 환경에서 장시간 노출되면 변형이 되며 건식 개질시 발생하는 탄소 침착으로 인해 빠르게 비활성화된다는 문제가 있다[3]. 또 다른 방법은 플라즈마를 사용하는 방법인데, 플라즈마는 활성종들을 통해 반응에 필요한 활성화 에너지를 쉽게 극복하게 하는 있다는 장점이 있다. 특히, 열 플라즈마는 고온의 환경을 조성할 수 있어 비열 플라즈마에 비해 높은 개질율과 에너지 효율을 보여주어 최근 많은 연구가 진행되고 있다[4].
공정의 최적화를 위해서는 실험과 함께 시뮬레이션과 검증이 필수적이며, 이를 위해서는 적절한 반응 메커니즘을 사용하는 것이 중요하다. 현재 열 플라즈마를 이용한 저분자 탄화수소 건식 개질의 시뮬레이션을 검증하는 데 주로 Gri-Mech 3.0이 사용되고 있다[5]. 하지만, Gri-Mech 3.0은 약 1000-2500 K 온도 범위에서 메탄 연소에 최적화된 메커니즘으로, 열 플라즈마와 같은 고온 분위기에 활성화되는 반응 경로가 누락되었을 가능성이 있다. 또한, Gri-Mech 3.0은 프로판에 대해 제한된 범위의 반응만을 포함하고 있어, 프로판을 제외한 C3 계열 탄화수소의 농도와 기여를 평가하는 데 어려움이 있다. 그러므로, 고온 환경에서 저분자 탄화수소 건식 개질 반응 해석에 사용 가능한 반응 메커니즘의 제시가 필요하다.
수십 개의 화학종과 수백 개의 반응들로 구성된 상세 반응 메커니즘을 수작업으로 만드는 것은 비용과 시간이 많이 소요되며, 오류가 발생할 가능성이 크다. 최근에는 이러한 문제를 해결하기 위해 reaction mechanism generator(RMG)라는 자동 반응 메커니즘 생성 도구가 등장하였다[6]. RMG는 Massachusetts Institute of Technology(MIT)에서 개발된 Python 기반의 오픈 소스 소프트웨어로, 반응 네트워크를 생성하여 특정 온도, 압력에서 초기 반응물로부터 생성될 수 있는 모든 화학종을 고려한 후, 반응 상수와 열역학적 매개변수를 추정하여 반응에 주요한 화학종과 반응들로 구성된 메커니즘을 자동으로 구축한다. 또한, RMG는 탄화수소 산화와 관련한 데이터베이스를 풍부하게 제공하여 열분해 및 연소 반응 메커니즘을 생성하는 데 특화되어 있다.
따라서, 본 연구에서는 RMG를 활용하여 플라즈마 에탄 건식 개질이 활성화되는 고온 범위에 사용 가능한 상세 반응 메커니즘을 생성하였다. 이때, Gri-Mech 3.0에서 고려되지 않은 C3 계열 탄화수소를 포함시켜 반응 중간체의 영향을 분석하였다. 메커니즘을 검증하기 위해 마이크로웨이브 열플라즈마 반응기 형상을 고려한 수치해석 모델링을 수행하였으며, 반응 경로 분석을 통해 두 메커니즘의 차이를 비교하고 Gri-Mech 3.0의 타당성을 검토하였다.
2. 방법론
2.1 RMG의 반응 메커니즘 생성
RMG의 열역학적 매개변수 추정 방식은 화학종에 대해 알려진 열역학적 매개변수가 데이터베이스에 존재하면 우선적으로 그 값을 사용하고, 만약 값이 데이터베이스에 없으면 Benson의 group additivity 방식[7]으로 추정하게 된다. 이 방식은 양자 화학 계산보다는 신뢰도가 낮지만 비교적 빠른 시간에 적절한 값을 계산할 수 있다는 장점이 있어 RMG에서는 기본적으로 이 방식을 통해 추정한다. 또한, group additivity 방식으로 추정한 값이 특정 화합물에는 다소 부정확할 수 있는데, 이 경우 on-the-fly semi-empirical 양자 화학 계산을 통해 더 정밀하게 값을 추정할 수도 있다.
반응 속도의 추정 방식은 기본적으로 reaction library에 저장되어 있는 반응 속도 상수를 우선으로 사용하며, 동일 반응이 reaction library가 참조하는 메커니즘들에 중복해 포함되어 있는 경우 나열한 순서 중 먼저 나타나는 메커니즘의 반응 속도 상수가 우선순위를 갖는다. 만약 RMG에서 도출한 반응이 reaction library에 없는 경우에는 “rate rule”에 의해 계층 트리(hierarchical tree)를 따라 추정하게 된다. RMG-database에는 수십 개의 rate family가 있으며 각각은 계층 트리 구조로 개별 반응 형태의 정보가 담겨있는 반응 레시피(reaction recipe)와 해당 반응 속도의 상수들을 포함한다. 계층 트리의 상위 노드는 포괄적인 반응으로 구성되고, 하위 노드로 내려갈수록 반응이 점차 구체화된다. RMG는 반응이 일치할 때까지 계층 트리의 하위 노드로 내려가 반응 속도를 추정하며, 만약 일치하지 않는다면 가장 가까운 노드(즉, 해당 이분자 반응과 가장 비슷한 화학종 쌍들의 반응)에 대응하는 반응들의 속도를 온도별 평균하여 속도 상수를 추정한다. 따라서, 반응 레시피가 없는 반응에 대한 반응 속도 상수는 부정확한 값을 갖게 되는데, RMG에서는 기존 계층 트리에 존재하지 않는 새로운 반응 레시피를 추가시켜 계층 트리를 수정하고 재학습시킴으로써 개선된 추정을 할 수 있도록 있다. 이때, 재학습에 필요한 반응 속도의 매개변수 데이터는 문헌 조사나 RMG의 automatic rate calculator(ARC)를 통해 구할 수 있다. 한편, 현재 RMG는 기상 반응만을 고려할 수 있으며, RMG에서 반응 속도는 수정된 Arrhenius 식 (1)으로 표현된다.
또한, 가역 반응의 경우 역방향 반응 속도는 평형 상수에 의해 계산되며 평형상수는 식 (2)과 같이, 깁스 자유 에너지를 통해 계산된다.
메커니즘은 Susnow의 rate-based 알고리즘[8]을 통해 생성되며, 이 방식에는 core와 edge라는 개념이 존재한다. Core는 최종 메커니즘에 포함될 화학종과 반응들이 포함되는 공간을 의미하며, edge는 RMG에서 고려하는 모든 반응과 화학종들이 존재하는 공간이다. 먼저, 사용자는 사용할 thermodynamic library와 reaction library의 데이터를 선택한다. 이때, reaction library에 있는 화학종과 반응들은 자동으로 중복을 감지하여 edge에 포함된다. RMG는 기본적으로 등온, 등압의 반응기(isothermal, isobaric reactor)를 사용하며 사용자는 우선적으로 반응기에 온도, 압력, 초기 화학종의 조성 및 몰분율, 그리고 종료 기준을 정한다. 초기 화학종들은 core에 들어가게 되며 이 화학종들로부터 발생할 수 있는 모든 반응들을 edge에 나열한다. 그런 다음 edge의 화학종 i들 중 매 시점 해당 화학종의 플럭스 Ri=dCi/dt가 ϵRchar를 초과하는 화학종들을 core에 추가한다. 여기서 ϵ은 사용자가 지정한 허용 오차이며 특성 플럭스인 Rchar은 식 (3)과 같이 계산된다.
Edge에 있는 화학종이 core에 새롭게 추가되면, edge에 있는 반응들 중에서 core에 속한 화학종들이 모두 포함된 반응들도 core에 새롭게 추가된다. 이러한 과정을 종료 기준을 충족할 때까지 반복하며, core 공간은 점차 확장한다. 최종적으로, 해당 화학종 간의 농도 변화에 크게 영향을 미치는 반응들로 메커니즘이 구성된다. 만약 온도, 압력이 다른 여러 개의 반응기들로 입력을 구성하여 반응 메커니즘 생성을 진행한다면, 알고리즘은 각각의 반응기에 대해 순차적으로 core 공간을 구성하고 확장하게 한다. 메커니즘 생성이 완료된 후, RMG는 핵심 반응에 포함된 모든 반응에 대해 물리적으로 불가능한 속도 상수가 있는지 판단하기 위해 충돌 속도 상수(collision rate constant)를 계산하고, 이를 토대로 속도 상수가 충돌 한계(collision limit)를 위반했는지 평가한다.
2.2 메커니즘 생성 모델 입력변수 설정
본 연구의 메커니즘 생성 모델은 RMG-Py(version 3.2.0)와 RMG-database(version 3.2.0)[9]를 사용하였으며, 총 4개의 등온, 등압의 반응기로 구성하였다. 초기 화학종은 모두 에탄, 이산화탄소, 그리고 아르곤(압력 의존 반응을 위한 불활성 기체)으로 설정하였다. 몰분율은 에탄, 이산화탄소, 그리고 아르곤에 대해 각각 0.09, 0.21, 그리고 0.70 (즉, 에탄-이산화탄소 혼합물 30%와 불활성 기체 70%)로 설정하였다. Gri-Mech 3.0을 바탕으로 한 시뮬레이션 결과로부터, 개질 반응은 플라즈마 온도인 5000 K부터 시작하여 약 1300 K에서 완료되었다. 따라서, 이를 고려해 등온, 등압의 반응기들의 온도는 각각 1500, 2500, 3500, 4500 K로 설정하였고, 압력은 모두 1 bar로 설정하였다. RMG에서는 생성되는 메커니즘이 과도하게 커지는 것을 방지하기 위해 제약 조건을 설정할 수 있다. 에탄 건식 개질의 가스 크로마토그래프 (GC) 분석 결과 대부분의 화학종들이 C2 이하의 화학종이었기 때문에, 본 연구에서는 C3 탄화수소들까지만 상세 반응 메커니즘의 화학종으로 고려될 수 있도록 설정하였다. 모델의 tolerance는 0.02로 설정하였고, 이는 RMG에서 권장하는 수치 범위인 0.01~0.05 내에 해당한다[10]. Thermodynamic library로는 저분자 탄화수소 반응 메커니즘인 USC-Mech-ii[11]을 수정한 라이브러리와, RMG의 group additivity 방식으로 추정된 열역학적 데이터를 포함한 primaryThermoLibrary를 사용하였다. 수정한 라이브러리는 USC-Mech-ii의 화학종 중에서 C3 이하의 화학종을 고려하였고, 이들 화학종들의 NASA 7-coefficient polynomials는 Burcat’s thermodynamic data[12]와 RMG-database를 참고하여 최대 온도 범위 5000 K 이상에서 사용 가능한 데이터들로 구성하였다. 이는 thermodynamic library에 있는 화학종의 온도 범위가 반응기 설정 온도 범위 밖에 있을 경우, 평형 상수 값 계산에 필요한 깁스 프리 에너지 계산이 불가능하여 역반응 속도를 계산할 수 없기 때문이다. Reaction library에는 RMG-database에 있는 저분자 탄화수소 메커니즘인 GRI-Mech3.0과 ERC-FoundationFuelv0.9, 그리고 combustion_core/version5을 사용하였고, seed mechanism은 사용하지 않았다. 추가적으로, 압력 의존 반응들의 중요성을 파악해보기 위해 모델에는 압력 의존 반응기가 포함된다. 압력 의존 반응기에서는 modified strong collision 근사 방법을 사용하여 마스터 방정식을 풀어 반응 속도 상수를 구한다[13]. 압력 의존 반응은 300 K에서 5000 K까지 8개의 구간, 0.01 bar에서 100 bar까지 6개의 구간에서 계산되며, 반응 속도는 6개의 온도, 4개의 압력 포인트에서 보간되어 Chebychev 다항식으로 표현된다. 마지막으로, RMG-Py(version 3.2.0)를 통해 생성된 메커니즘은 yaml 형식으로 저장되어 Cantera[14]의 입력값으로 별도 후처리 없이 사용될 수 있도록 하였다.
3. 연구 결과 및 고찰
최초 메커니즘 생성 시도 시, 반응 C2H5 + C3H4 → C2H6 + C3H3, C2H3 + C3H4 → C2H4 + C3H3, C3H4 + C3H5 → C3H3 + C3H6이 충돌 한계를 위반하였다. C2H5 + C3H4 → C2H6 + C3H3와 C2H3 + C3H4 → C2H4 + C3H3 반응은 Aäron G. Vandeputte가 group additivity 방식으로 추정한 탄화수소의 hydrogen abstraction 반응이었고[15], C3H4 + C3H5 → C3H3 + C3H6 반응은 RMG의 rate rule에 의해 추정된 값이었다. Aäron G. Vandeputte의 반응 속도 매개변수는 300-1500 K 범위에서 최적화된 값이었으나, 이를 높은 온도 범위에서 그대로 사용한 것이 충돌 한계 위반의 원인으로 추정된다. 이를 보완하기 위해, 보다 고온에서 최적화되어 있는 Creck modeling group의 C1-C3 상세 반응 메커니즘을 이용하였다[16]. 해당 메커니즘에는 최대 C10 화학종에 대해 group additivity 방식으로 추정한 탄화수소 hydrogen abstraction 반응들이 포함되어 있으며, 해당 3개의 반응에 대해 정반응과 역반응(총 6개 반응)이 모두 존재한다. 따라서, 이 6개 반응을 추출하여 모델을 재학습하였고, 그 결과 이어진 메커니즘 생성 시도 시에는 충돌 한계 위반이 더 이상 발생하지 않았다. 최종적으로, 메커니즘은 49개의 화학종 (Ar, He, Ne, N2, C2H6, CO2, O, H2, H, OH, CH, CO, CH2, HCO, CH2(S), CH3, CH2O, CH4, CH2OH, CH3O, C2H, C2H2, C2H3, C2H4 3종, C2H5, H2O, H2CC, C3H2, C3H3 2종, C3H4 3종, C3H5 4종, C3H6 3종, C3H7 2종, CHO2, C2H2O2 3종, C2H3O2)와 652개의 반응으로 구성되었으며, 부록의 웹저장소에 제공하였다. 비록 메커니즘은 몇 가지 온도의 등온, 등압 연소기에 대해 추출되었지만, 최종 추출된 메커니즘은 메커니즘 추출이 진행된 온도 범위의 주요 화학종과 반응경로를 모두 포함하게 되어 범용적으로 사용이 가능하다.
Kang 등의 연구[17]에서 대기압 마이크로웨이브 열플라즈마를 사용해 에탄 건식 개질 실험(즉, 반응기 출구에서의 화학조성 분석을 위한 GC 실험과 플라즈마 발생 영역 온도 측정을 위한 발광분광분석 실험)과 해석을 진행하였고, 해석은 Fig. 1과 같이 반응기 네트워크 모델로 구성하여 진행하였다. 구체적으로, 모델은 플라즈마 흐름과 우회 흐름 두 영역으로 구분되며, 각각 원통형과 중공원통형으로 간략화된다. 각 흐름은 축방향을 따라 연속적으로 반응이 진행되며, 동시에 두 흐름 계면에서는 확산을 통해 열 및 물질 교환이 이루어진다. 각 흐름은 2 cm 높이의 continuous stirred tank reactor(CSTR)들로 구성된 연속흐름반응기로 설계하였으며, 플라즈마 발생 위치로부터 총 62 cm 하류 지점을 포함한다. 본 연구에서는, RMG를 통해 생성한 상세 메커니즘 검증을 위해, Kang 등의 연구[17]과 동일하게 동일한 반응기 형상 하에서 플라즈마 출력 2 kW, 총 유량 10-25 LPM 조건에 대해 반응기 네트워크 시뮬레이션을 수행하였다. 반응기 네트워크 모델은 Python 기반 Cantera 패키지를 사용하여 구현되었으며, 사용한 상세 반응 메커니즘을 제외한 반응기 모델의 나머지 값들은 Kang 등의 연구 [17]에서 사용된 것과 모두 동일하다. 반응기 모델의 보다 자세한 가정과 수식들은 Kang 등의 연구[17]에 소개되어 있다.
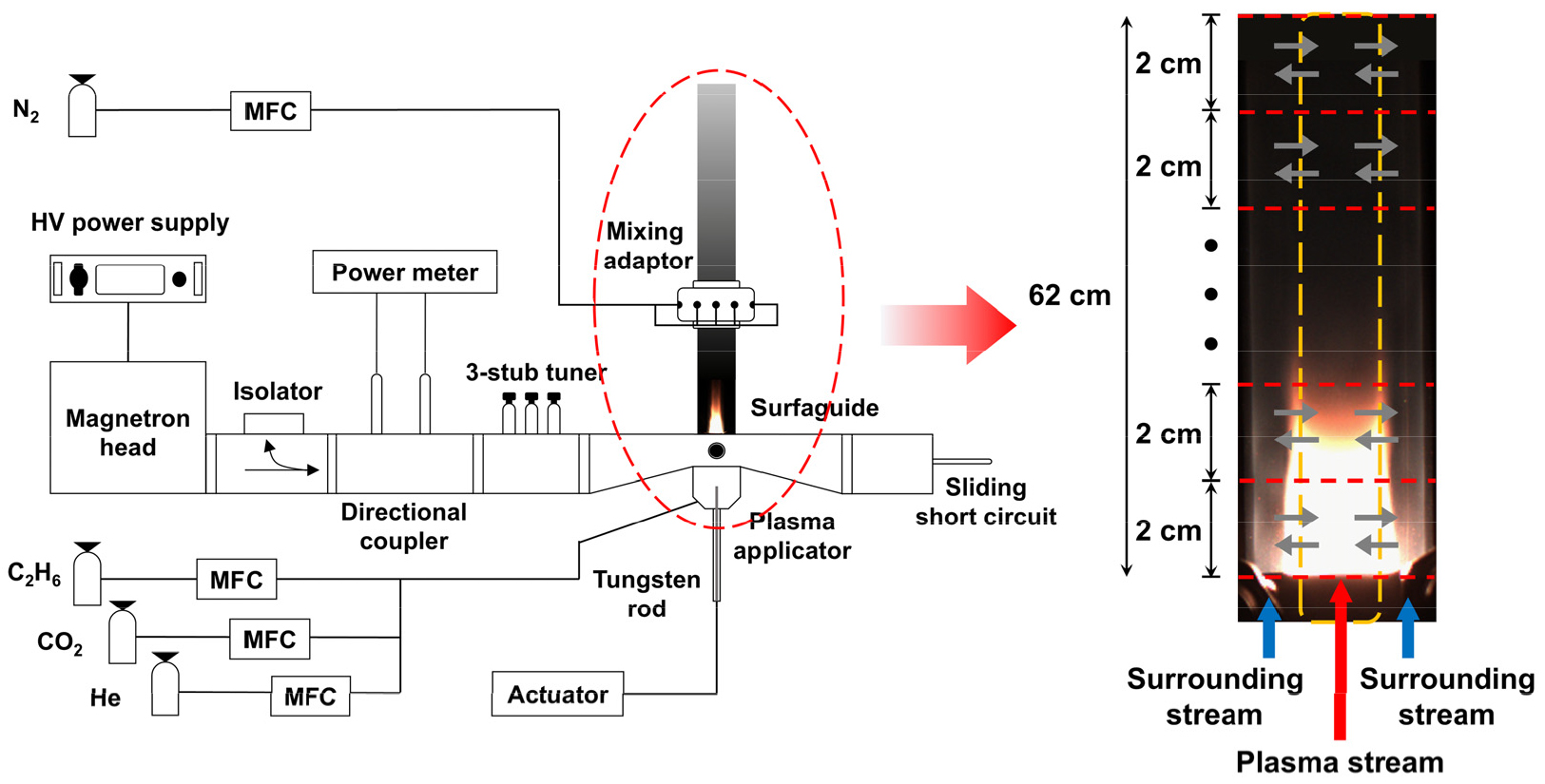
Fig. 2는 GC 측정과 RMG를 사용해 생성한 상세 메커니즘을 적용해 획득한 시뮬레이션 몰 농도의 비교 결과를 보여준다. Fig. 2(a)와 Fig. 2(b)는 각각 주요 화학종 CO, H2, CO2, 그리고 C2H6와 반응 중간체인 CH4, C2H2, 그리고 C2H4의 GC 측정과 시뮬레이션 몰 농도를 비교한 결과를 보여준다. H2O는 GC 측정에서 ghost peak를 유발하므로 실험에서는 제습제에 의해 물이 제거되어 H2O의 측정 결과는 없고, 시뮬레이션의 H2O 몰 농도만 Fig. 2(b)에 나타나 있다. Fig. 2(c)는 aC3H4, pC3H4, C3H6의 시뮬레이션 몰 농도로, Gri-Mech 3.0에 포함되지 않은 C3 계열의 화학종을 나타낸다. 총 유량 10 LPM에서 CO, H2, CO2, C2H6의 시뮬레이션 몰 농도는 각각 46.25, 35.35, 7.24, 그리고 0.00%였으며, 총 유량 25 LPM에서 CO, H2, CO2, C2H6의 시뮬레이션 몰 농도는 각각 25.16, 21.16, 33.33, 그리고 11.37% 였다. 시뮬레이션 결과에는 H2O의 몰 농도가 반영되어 있지만 실제 GC 측정 결과에는 H2O가 빠져 있으므로, 이를 감안한다면 시뮬레이션이 실험 결과를 잘 예측하고 있음을 알 수 있다. 총 유량 10 LPM에서 CH4, C2H2, C2H4, H2O의 시뮬레이션 몰 농도는 각각 1.93, 4.44, 0.37, 그리고 4.39%였고, 총 유량 25 LPM에서 CH4, C2H2, C2H4, H2O의 시뮬레이션 몰 농도는 각각 3.05, 1.75, 2.47, 그리고 1.50%였다. 한편, RMG에서 생성한 메커니즘은 Gri-Mech 3.0에 포함되지 않은 화학종들을 고려하기 때문에, 특히 C3 탄화수소들을 포함한 다양한 부산물들을 결과에 포함할 수 있다. 그럼에도 불구하고, Kang 등의 연구[17]에서 확인되었듯이, aC3H4, pC3H4, C3H6의 시뮬레이션 몰 농도는 모든 유량 구간에서 거의 발견되지 않았다. 한편, C2 탄화수소의 몰 농도는 실제 GC 측정 결과보다 최대 3% 과대 예측되었으며, 이는 Gri-Mech 3.0로 예측한 결과와 유사하였다. 즉, 이러한 반응 중간체(intermediates)의 과대 예측은 반응이 누락되어 발생한 것이 아니라, 유동을 플라즈마 흐름과 우회 흐름의 두 흐름들로 간략화한 모델의 단순성에서 기인하는 것으로 사료된다.
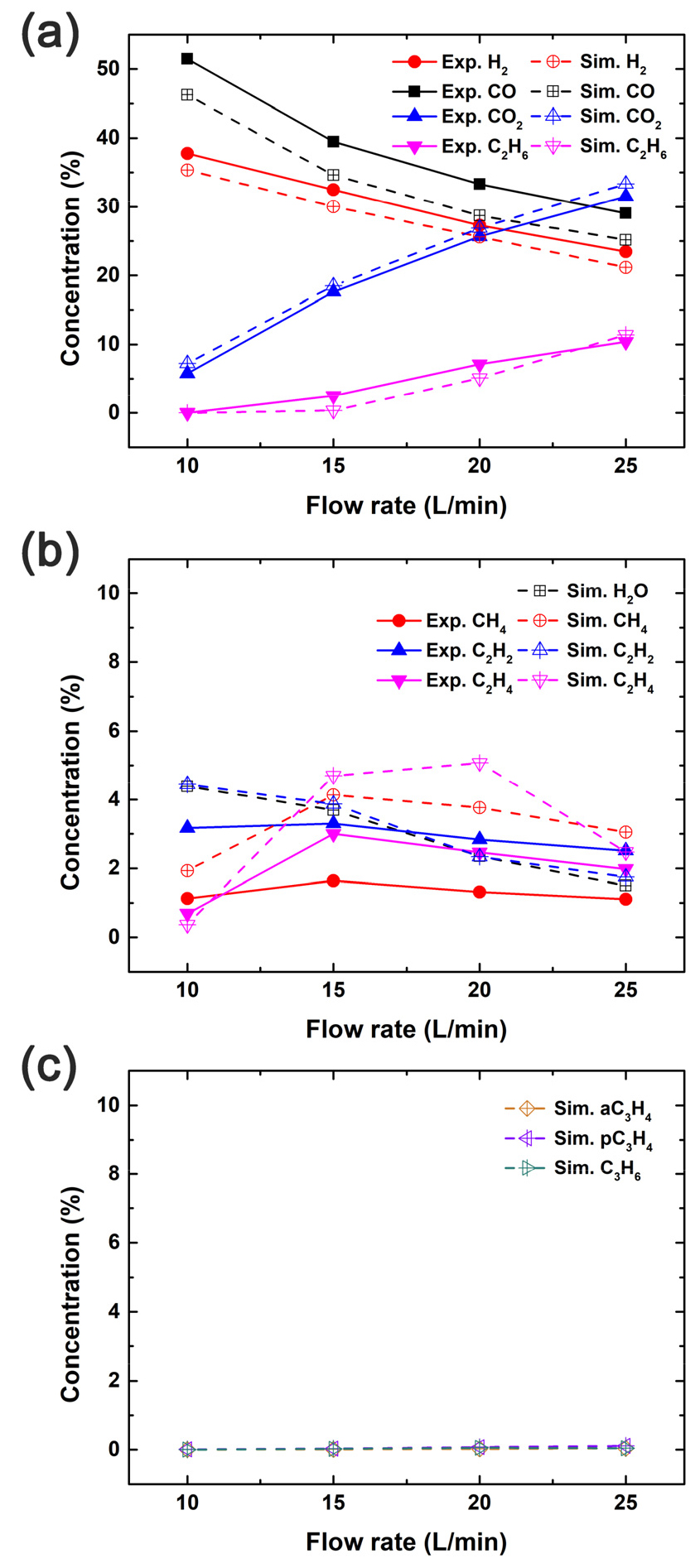
Fig. 2.
Comparison between the calculated and measured mole fractions of (a) H2, CO, CO2, and C2H6, and (b) CH4, C2H2, and C2H4 at 0.61 m downstream from the Surfaguide as a function of the C2H6-CO2 mixture flow rate. In (b) and (c), simulated H2O, aC3H4, pC3H4 and C3H6 concentrations are also displayed.
RMG로 생성한 메커니즘을 사용해 진행한 마이크로웨이브 열플라즈마 에탄 건식 개질의 더 자세한 과정을 파악해보기 위해 반응 경로 분석을 수행하였다. 각 흐름의 CSTR 반응기의 높이는 2 cm였고 시뮬레이션에서 surfaguide 하류 16 cm 이후에서 합성가스로의 전환은 거의 마무리되었으므로, 총 9개의 반응기의 반응 속도를 합산하여 계산하였다. 이때, 반응 속도는 플라즈마 흐름과 우회 흐름의 서로 다른 CSTR 반응기 부피를 고려하여 식 (4, 5)로 계산되었다.
각각 j는 반응기 순번, m은 반응 순번, Ai는 화학종 i의 몰농도, νi와 νi'는 각각 화학종 i에 대한 반응물과 생성물의 화학양론 계수, N은 전체 화학종의 수를 나타낸다. Vp와 Vs는 각각 플라즈마 흐름과 우회 흐름을 구성한 개별 CSTR 반응기의 부피를 의미한다.
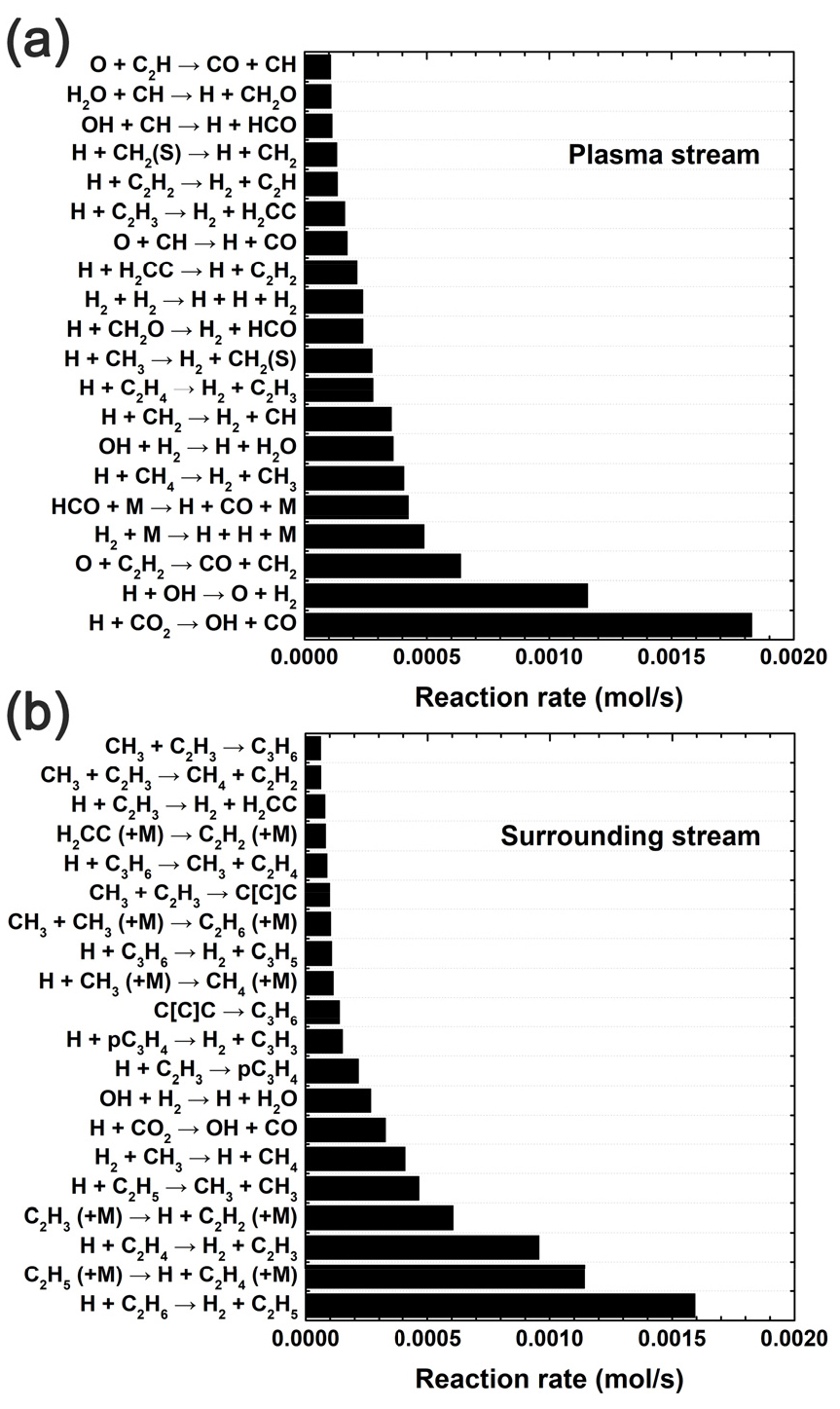
Gri-Mech 3.0을 이용한 반응 경로 분석 결과는 Kang 등의 연구[17]에 제시되어 있으며, Fig. 3은 총 유량 10 LPM 조건에서 RMG로 생성한 메커니즘을 적용해 획득한 결과로부터 도출한 주요 반응 경로 분석 결과를 보여준다. 수백 개의 반응들의 반응속도를 모두 나열하는 대신, 두 흐름에서 반응 속도가 가장 빠른 상위 20개의 반응들을 정렬하여 제시하였다. 먼저 Fig. 3(a)에 나와 있듯이, 플라즈마 흐름에서 CO의 주요 생성 경로는 CO2 + H → CO + OH가 가장 지배적이었고, O + C2H2 → CO + CH2, HCO + M → H + CO + M, O + CH → H + CO, O + C2H → CO + CH를 통해 CO가 생성되었다. 즉, RMG로 생성한 메커니즘은 라디칼 H와 O를 CO 생성의 핵심 화학종으로 예측하였다. H2의 주요 생성 경로는 H + OH → O + H2 반응과 함께, H + CH4 → H2 + CH3, H + CH2 → H2 + CH, H + C2H4 → H2 + C2H3, H + CH3 → H2 + CH2(S), H + CH2O → H2 + HCO, H + C2H3 → H2 + H2CC, H + C2H2 → H2 + C2H과 같이 C2 이하 탄화수소의 수소 추출 반응이었다. 따라서, 라디칼 H와 OH가 H2 생성의 핵심 화학종임을 알 수 있다. Gri-Mech 3.0을 사용해 예측한 개질 경로 분석 결과와 RMG로 생성한 메커니즘을 사용해 예측한 개질 경로 분석 결과가 대체로 동일하였고, 차이점으로는 Gri-Mech 3.0은 플라즈마 흐름에서 화학종 C를 포함한 반응들을 주요 반응 경로로 예측한 반면 RMG에서는 화학종 C와 C가 포함된 반응들을 core에 포함시키지 않았다는 점이다. 해당 실험 조건들에서 고형 탄소를 포함한 미측정 물질들의 질량 분율이 적었다는 것을 고려하면, 이는 실험 결과를 잘 반영하는 것을 시사한다.
한편, Fig. 3(b)는 우회 흐름에서의 반응 경로 분석 결과를 보여준다. 상위 20개의 반응 중 10개가 중간 반응체 형성과 관련된 압력 의존 반응인 것으로 나타났다. 이는 우회 영역의 온도 범위인 약 1000-1500 K에서 압력 의존 반응이 특히 활성화되며, 중간 반응체 농도의 정확한 예측을 위해 압력 의존 반응을 고려하는 것이 중요하다는 것을 보여준다. CO는 CO2 + H → CO + OH 반응을 통해 생성되었으나, 플라즈마 흐름에서의 생성 속도에 비해 상대적으로 낮은 속도였다. H2는 H + C2H6 → H2 + C2H5, H + C2H4 → H2 + C2H3, H + pC3H4 → H2 + C3H3, H + C3H6 → H2 + C3H5, H + C2H3 → H2 + H2CC와 같이 C2 이상 탄화수소에서 수소 추출 반응을 통해 생성되었다. 플라즈마 흐름에서는 온도가 충분히 높아 더 짧은 탄화수소로 분해되는 반면, 우회 흐름에서는 상대적으로 낮은 온도로 인해 긴 탄화수소의 분해 반응이 주로 일어남을 알 수 있다. 또한, C3 탄화수소를 포함한 반응을 자세히 살펴보면, C3 탄화수소들은 우회 흐름에서 H + C3H3 → pC3H4, CH3 + C2H3 → C[C]C, CH3 + C2H3 → C3H6, C[C]C → C3H6 등의 재결합과 이성질화 반응을 통해 주로 생성되었지만, 동시에 H + C3H6 → CH3 + C2H4, H + pC3H4 → CH3 + C2H2와 같은 반응을 거쳐 C2 이하의 탄화수소로 다시 분해되었다(속도가 빠른 상위 20개 반응에는 미포함). 결과적으로, surfaguide 후단 62cm 지점에서는 C3 탄화수소가 거의 발견되지 않았다. 이는 우회 흐름의 온도가 플라즈마 흐름 온도에 비해 상대적으로 낮지만, 여전히 온도가 높기 때문에 C3 탄화수소의 생성되기 어렵다는 것을 보여준다.
4. 결 론
본 연구에서는 고온 및 C3 이하의 탄화수소 건식 개질 해석에 사용 적합한 상세 반응 메커니즘을 RMG를 생성하고 Gri-Mech 3.0 사용 결과와 비교 분석하였다. RMG의 반응 메커니즘은 고온의 열플라즈마 개질 조건에 사용가능하도록 1500-4500 K 넓은 온도 범위에 대해 진행되었고, 압력 의존 반응의 영향을 반영하기 위해 압력 의존 반응기를 포함시켰다. 최종 상세 메커니즘은 49개의 화학종과 652개의 반응으로 구성되었다. 마이크로웨이브 플라즈마 에탄 개질에 대한 반응기 네트워크 모델에 적용하여, 두 메커니즘(Gri-Mech 3.0과 RMG로 생성한 메커니즘)의 예측 성능을 비교한 결과, 두 메커니즘 모두 실험적으로 측정한 주요 가스들의 농도값을 잘 예측하였다. 반응 중간체들의 경우, RMG로 생성한 메커니즘이 Gri-Mech 3.0에 포함되지 않은 C3 탄화수소들을 대거 고려했음에도, 실험 결과와 동일하게 C3 탄화수소들은 거의 생성되지 않는 것으로 잘 예측하였다. 여전히 C2 계열 탄화수소를 다소 과대 예측하였으나, 이는 반응기 네트워크 모델의 단순성에 기인한 것으로 판단된다. 추가적으로, 자세한 반응 과정을 파악하기 위해 플라즈마 흐름과 우회 흐름에 대해 반응 경로 분석을 수행하였다. 플라즈마 흐름에서 RMG로 생성한 메커니즘으로 예측한 주요 반응 경로는 Gri-Mech 3.0로 예측한 주요 반응 경로와 대체로 일치하였다. 한편, RMG로 생성한 메커니즘으로 예측한 결과로써, 우회 흐름에서는 C3 탄화수소의 중간체들이 압력 의존 반응으로 생성되었으나, 빠르게 C2 이하의 탄화수소로 분해되었다. 이는 우회 흐름의 온도가 C3 탄화수소의 생성되어 잔류하기에는 여전히 높은 온도 범위임을 보여준다. 마지막으로, Gri-Mech 3.0이 RMG로 생성한 메커니즘의 주요 개질 반응들을 대부분 포함하고 있어, Gri-Mech 3.0이 열플라즈마를 사용한 저분자 탄화수소 건식 개질 해석에 유효하게 사용될 수 있었음을 확인할 수 있었다.
